Lipid basics
Lipids are fatty substances that are required for maintenance of normal bodily function. Cholesterol and triglycerides are the major lipids that circulate in blood plasma and are transported in globules known as lipoproteins. Cholesterol is an important component of cell membranes and is required for the synthesis of steroid hormones and bile acids. As the daily requirement for cholesterol cannot be met from dietary intake, the majority (80%) is derived from biosynthesis in the liver. Triglycerides are the storage form of long chain fatty acids, derived from the diet or synthesised in the liver, which are an important source of energy and structural fatty acids required for formation of phospholipids, an essential component of cell membranes. Both the liver and the gut package cholesterol, triglycerides and fat-soluble vitamins into lipoproteins for delivery to other tissues.
Lipoprotein fractions
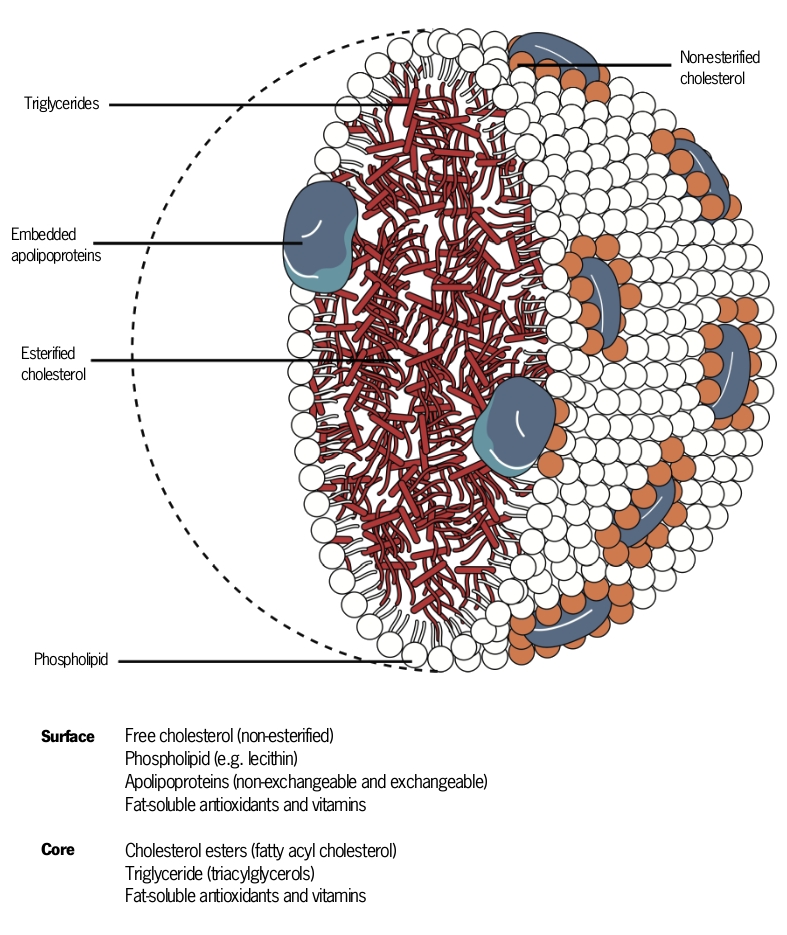
All lipoproteins have a common basic structure (figure 1) but they vary greatly in their size, density and composition (figure 2 and table 1).
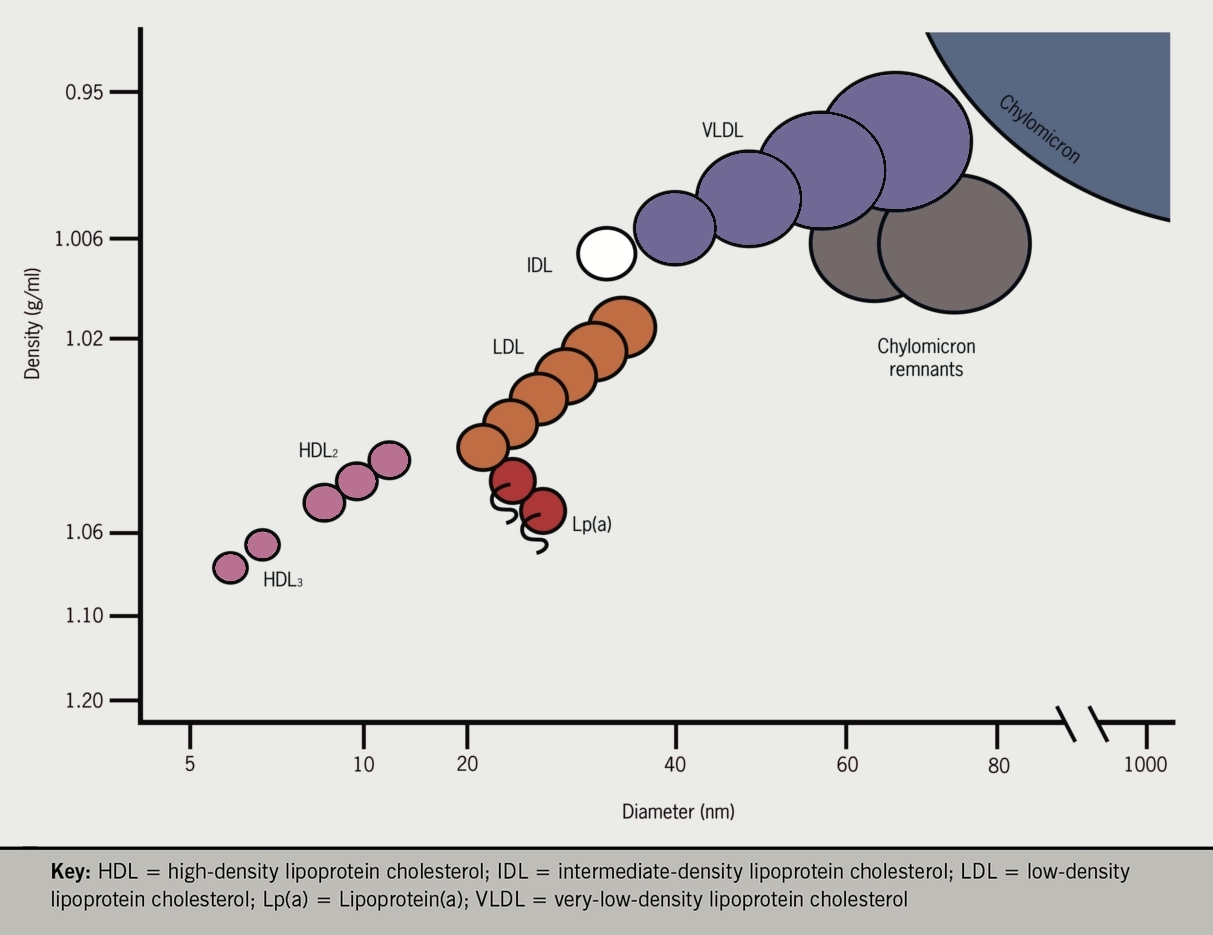

Lipid fractions are mixtures of lipoproteins of similar size and density which can be separated by the ultracentrifuge and are named accordingly as:
- high density lipoproteins (HDL)
- low density lipoproteins (LDL)
- very low density lipoproteins (VLDL).
As VLDL are metabolised into LDL (see below), short-lived intermediate density lipoproteins are also seen (IDL).
Chylomicrons, which appear after meals, are the largest and lowest density lipoproteins and rapidly float to the top of stored plasma without ultracentrifugation. Chylomicrons are rapidly metabolised into smaller VLDL sized particles (chylomicron remnants).
Apolipoproteins
In addition to their lipid components, lipoproteins contain specific protein components known as apolipoproteins which provide a structural framework and have a number of other important functions, including binding to receptors and activation of lipid transporters and metabolising enzymes. More detail on specific apolipoproteins can be found in the drop down box.
Specific apolipoproteins
Apolipoprotein B100 is the bulk carrier of endogenously produced lipids and is secreted by the liver as the major apolipoprotein component of VLDL and LDL, one molecule per lipoprotein, which stays with the particle until it is removed from the circulation by the LDL receptor. A shorter form of this apolipoprotein, apolipoprotein B48, is secreted by the intestine as the major structural component of chylomicrons which carry dietary (exogenous) lipids.
The large, triglyceride rich VLDL and chylomicrons also contain multiple copies of small exchangeable apolipoproteins (apolipoproteins AI AII AIV, CI CII CIII and E) which enter the HDL fraction as triglyceride is removed by triglyceride metabolising enzymes lipoprotein lipase (LPL) and hepatic lipase (HL). Apolipoprotein A1 is secreted by both the gut and the liver and is the major apolipoprotein component of HDL which has an important role in the reverse transport of cholesterol back to the liver from peripheral tissues.
Plasma lipid pathways
The three major pathways are co-ordinated by the liver to ensure that balance (homeostasis) of the major lipid classes is maintained, avoiding the twin perils of deficiency and overload:
- endogenous
- exogenous
- reverse.
Intestinal (exogenous) pathway
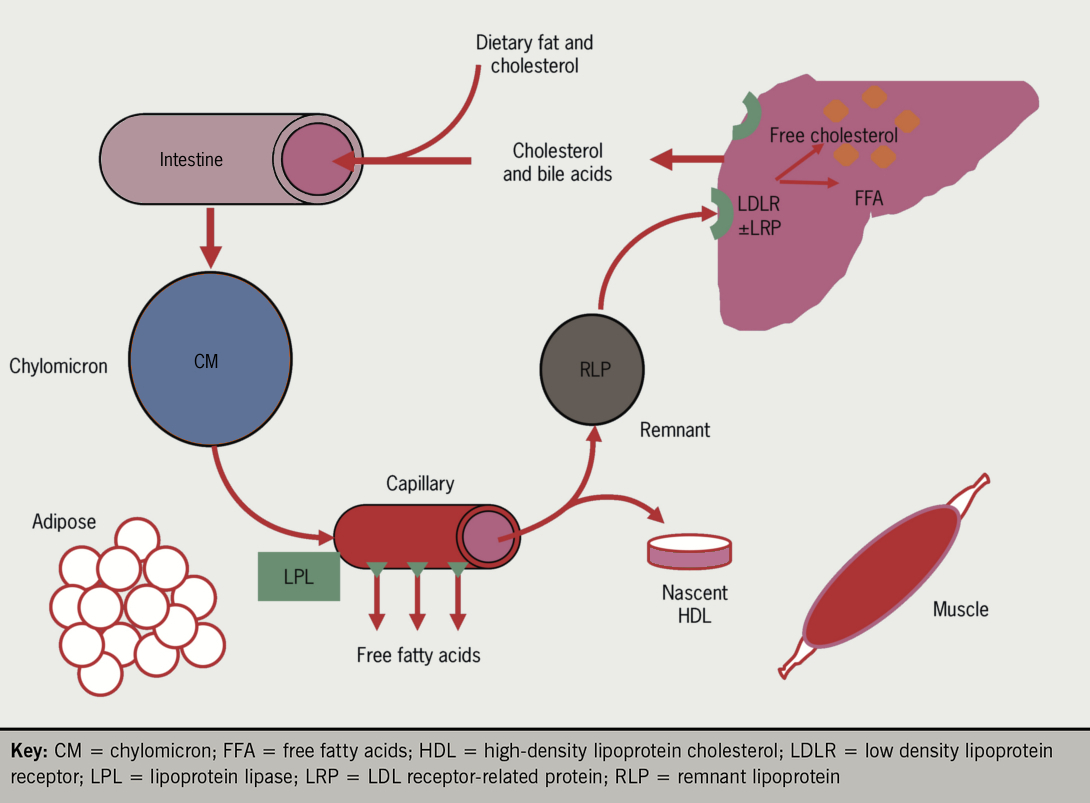
Figure 3 shows the intestinal (exogenous) pathway. Cholesterol and triglycerides derived from dietary lipids are absorbed in the gut and incorporated into chlyomicrons, regulated by the Niemann-Pick C1-Like 1 (NPC1L1) transporter and microsomal triglyceride transfer protein (MTP). These very large, triglyceride-rich lipoproteins are secreted into the lymph and bypass the liver, entering the plasma post-prandially via the thoracic duct. Chylomicrons deliver the fat to adipose tissue via lipoprotein lipase (LPL) which allows it to be taken up rapidly in the form of fatty acids. Once small enough the resulting chylomicron remnant is removed from plasma by the liver via apoE (see drop down box) binding to the remnant receptor (LRP) or to the LDL receptor (LDLR). Left over surface material and exchangeable apolipoproteins (AI AII AIV, CI CII CIII and E) may enter the HDL pool as ‘nascent HDL’. Between meals (post-absorptive phase), as insulin levels fall, fatty acids are released from adipose tissue by lipolysis (hormone sensitive lipase and adipose tissue triglyceride lipase) and enter the circulation where they are rapidly bound to albumin.
Hepatic (endogenous) pathway
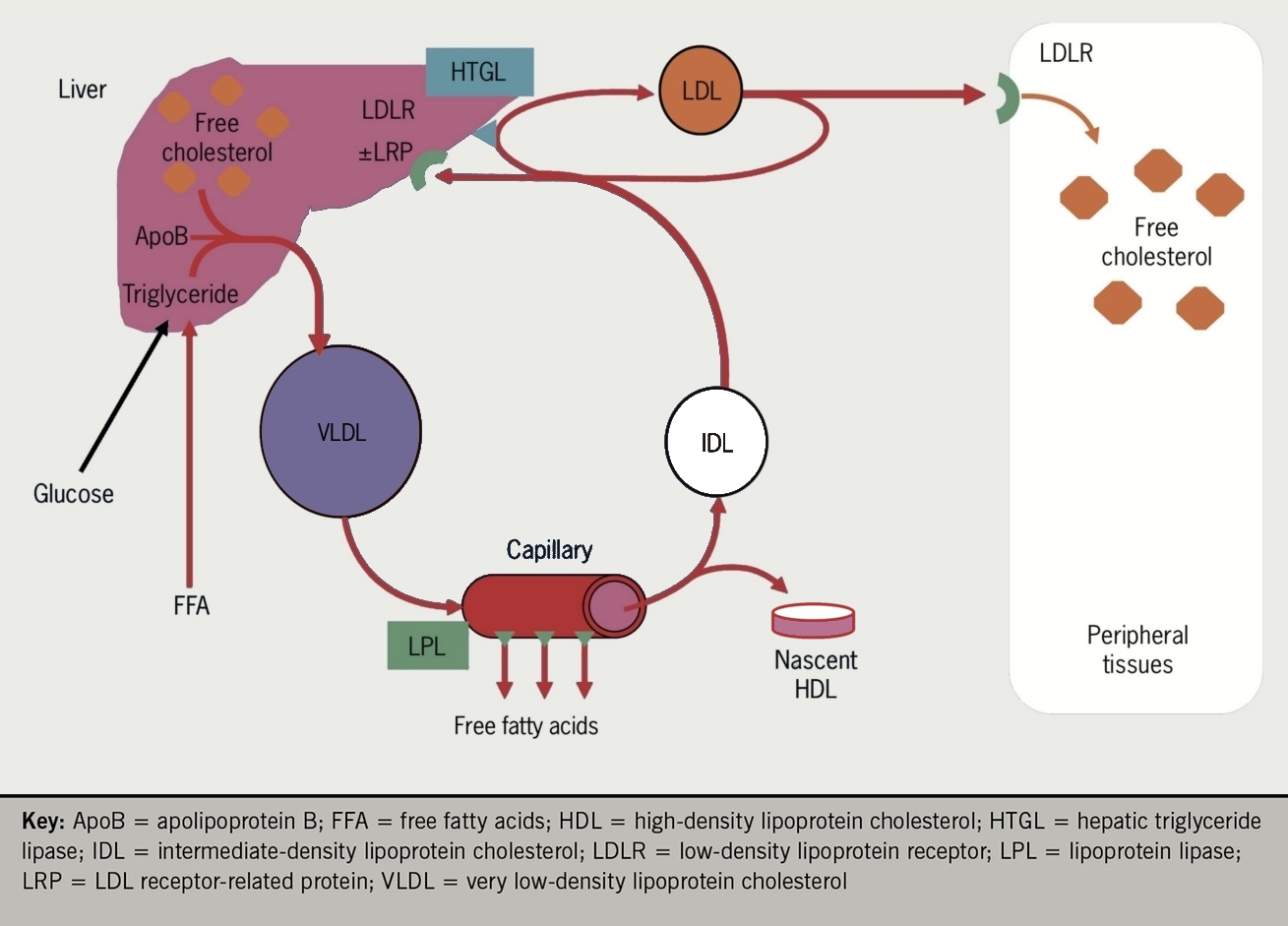
Fatty acids, arriving at the liver bound to albumin or newly-synthesised (de novo lipogenesis), are re-esterified to form triglyceride and together with cholesterol are loaded onto apoB to form VLDL. These large triglyceride rich lipoproteins enter the plasma between meals and deliver the fat to adipose tissue and muscle via lipoprotein lipase (LPL) which allows it to be taken up in the form of fatty acids (see figure 4). Once small enough, the resulting IDL is either taken up directly by the liver (cf. Chylomicron remnant) or converted to LDL by hepatic lipase (HL). The resulting LDL may then be taken up by peripheral tissues via the LDLR to meet local cholesterol needs. Any surplus LDL particles are finally removed by the liver via the LDLR.
Reverse cholesterol pathway
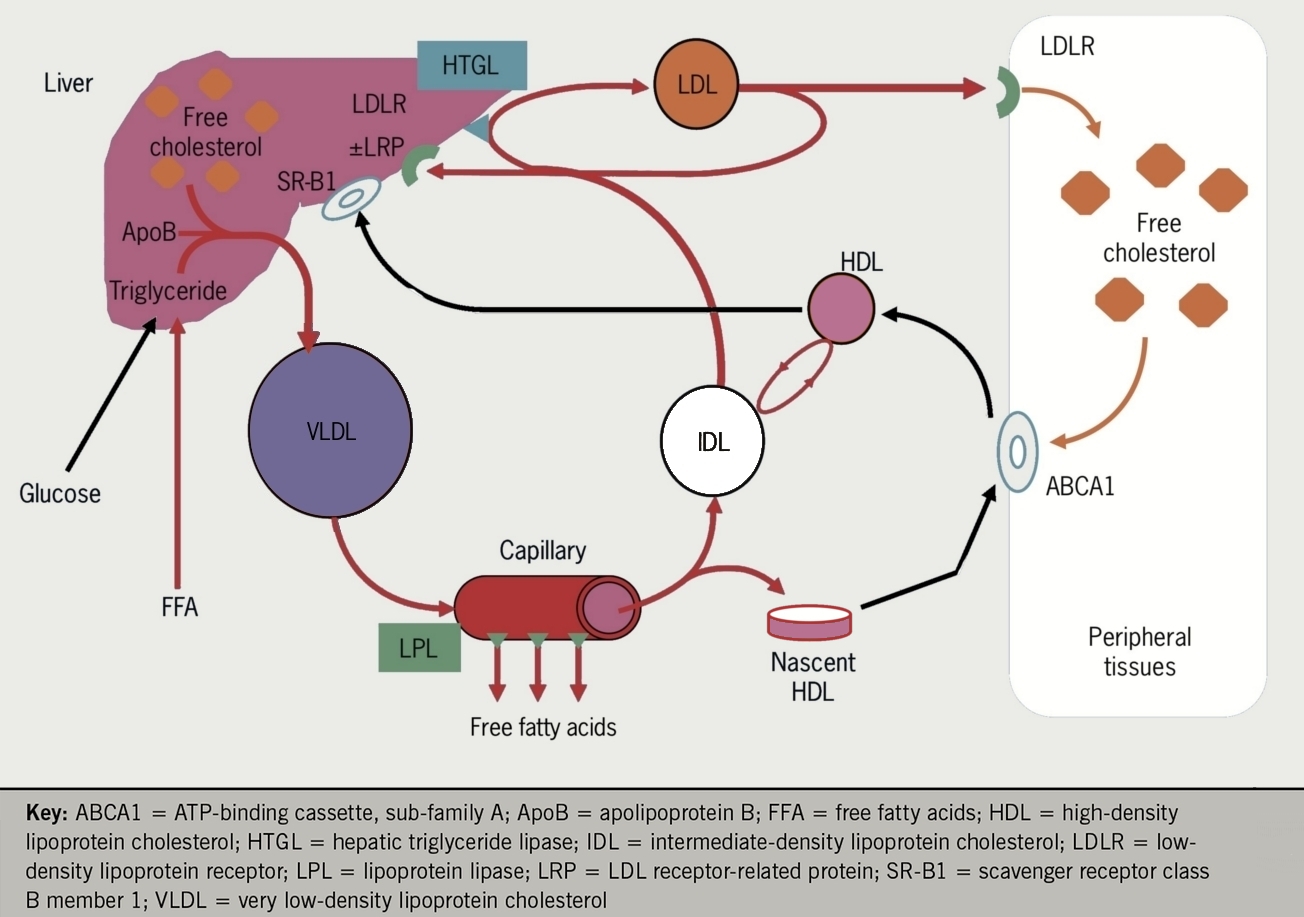
HDL inhibits the development of atheroma and coronary artery disease by transporting excess tissue cholesterol to the liver, where it is converted into bile acids and excreted (figure 5). Lower levels of HDL have been correlated with an increased risk of atherosclerosis, the primary cause of cardiovascular disease. The principal HDL pathway, termed reverse cholesterol transport (RCT) is a major contributor to lipid homeostasis. Cholesteryl ester transfer protein (CETP) is responsible for the exchange of cholesteryl esters from HDL for triglyceride in more atherogenic cholesterol fractions including LDL and VLDL. Some individuals with loss of function in the gene encoding CETP appeared to be at lower cardiovascular risk and CETP therefore became an attractive target for pharmaceutical inhibition.
Nascent HDL, in the form of flattened discs, is generated from LPL- mediated lipolysis of triglyceride rich lipoproteins (TRLs,) including (VLDL and cChylomicrons), or triglyceride rich lipoproteins (TRLs) or secreted directly from the gut or liver, and enters plasma and where it picks up additional exchangeable apolipoproteins.
Free cholesterol is removed from peripheral tissues (including cholesterol laden macrophages in the arterial wall) via ATP-binding cassette A1 (ABCA1) activated by apoA1, rapidly esterified by the action of lecithin cholesterol acyl transferase (LCAT) and funnelled into the core of the new HDL particle converting it to the mature spherical form. Core lipid exchanges between HDL and TRLs occur in the circulation, catalysed by CETP. This allows cholesterol to be offloaded from HDL into VLDL and LDL which are destined for hepatic uptake, permitting further cholesterol uptake from tissues. Finally the cholesterol-enriched HDL particle returns cholesterol to the liver via the scavenger receptor B1 (SCARB1) for biliary excretion. The cholesterol-depleted HDL particles can then return to the circulation to undertake more reverse cholesterol transport.
Cholesterol homeostasis
The rate of cholesterol formation by the liver and absorption by the small intestine is highly responsive to the cellular level of cholesterol. This feed back regulation is controlled primarily by changes in the amount and activity of 3-hydroxy-3 methylglutaryl CoA reductase (HMGCoA reductase). This enzyme catalyses formation of mevalonate, the committed step in cholesterol biosynthesis. (For more detail on this process of cholesterol homeostasis, see dropdown box).
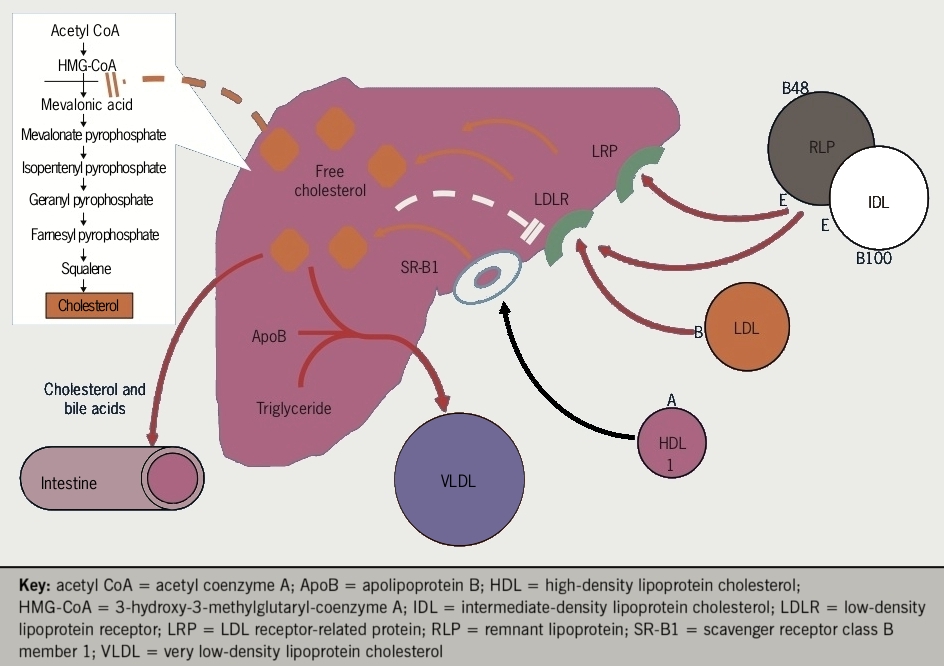
Cholesterol homeostasis
The concentration of free cholesterol determines the fluidity and function of cell membranes and regulates overall cholesterol homeostasis (see figure 6). The concentration of free cholesterol is ‘sensed’ by membrane bound transcription factors, known as sterol regulatory element binding proteins (SREBPs). When hepatic cholesterol content is reduced by export in lipoproteins or conversion to bile acids, membrane cholesterol concentration falls and SREBP-2 activates the enzymes of cholesterol synthesis, including HMG-Co-A reductase which is the rate limiting step in the pathway. SREBP-2 also activates the synthesis of LDL-receptors, accelerating the uptake of cholesterol in LDL to help restore membrane cholesterol concentration. In addition, SREBP-2 activates synthesis and secretion of proprotein convertase subtilisin/kexin type-9 (PCSK9), a counterbalancing factor which ‘applies the brakes’ by binding LDL-receptors and directing them to destruction in the lysosome, thereby preventing their recycling to the cell membrane.
Conversely, when hepatic cholesterol is increased by receptor mediated uptake of cholesterol in lipoproteins or return of cholesterol to the liver by HDL particles, membrane cholesterol increases, preventing activation of SREBP-2 and leading to LDL-receptor downregulation and inactivation of cholesterol synthesis.
Lipoproteins and atherogenesis
The majority of circulating cholesterol is carried in LDL which is the lipoprotein most closely associated with the development of atherosclerosis.

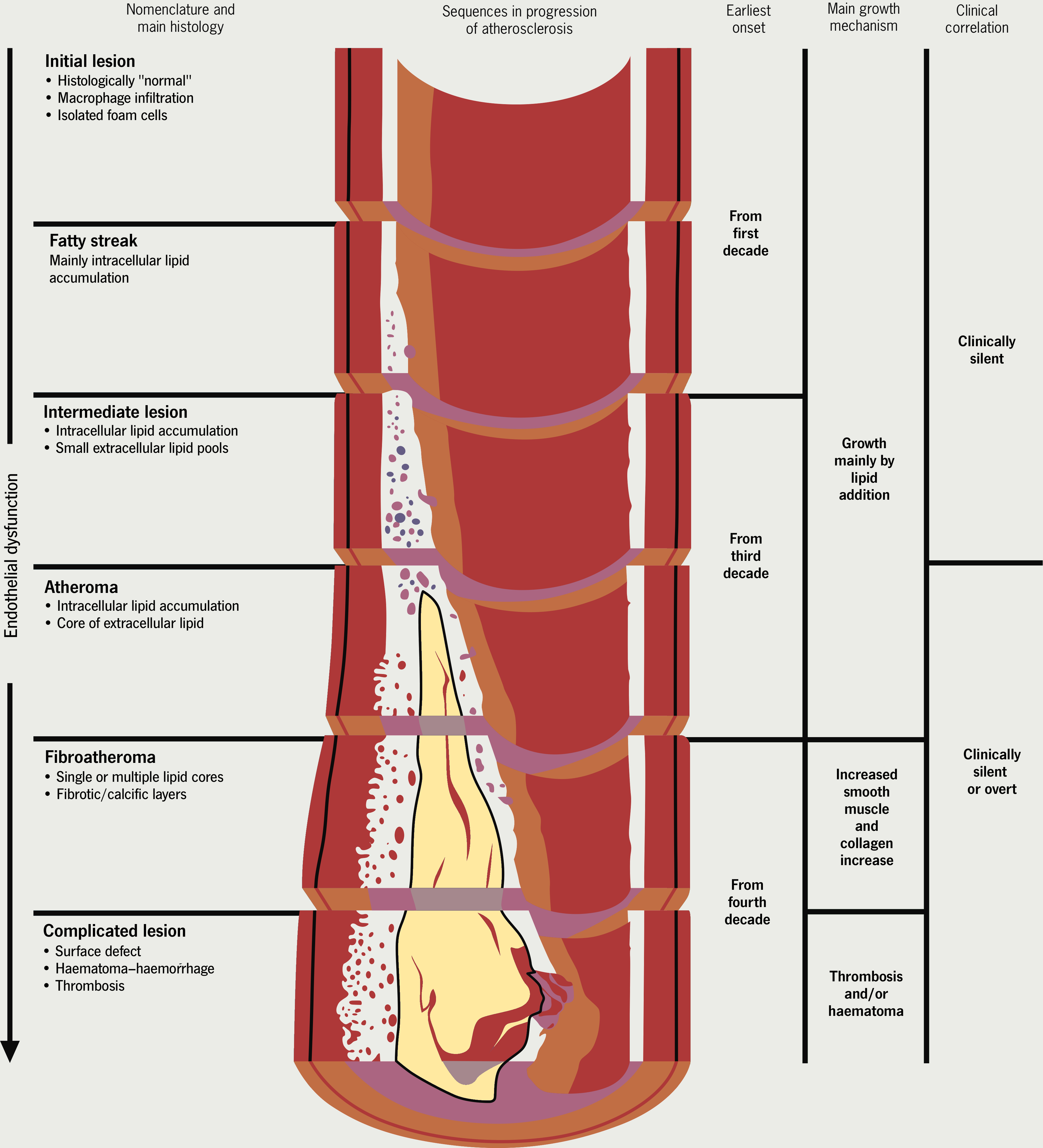
The steps in atherogenesis are summarised in table 2.
Under normal circumstances, LDL may pass from the plasma into the subendothelial space and return to the liver to be removed from the circulation. At this point it has performed its transport functions without being taken up by macrophages and indeed is unable to stimulate foam cell formation in vitro. However, if retention of the LDL in the endothelial space is increased, due to endothelial injury (e.g. with smoking, hyperglycaemia, hypertension) or if removal of LDL from the circulation is delayed, it can become damaged by oxidation or modified in other ways.
Oxidised or otherwise modified LDL are retained in the subendothelial space and are taken up by monocyte-derived macrophages via the scavenger receptor leading to the formation of foam cells, and the development of arterial sub-endothelial fatty streaks, the precursor of atheroma. Small dense LDL particles, typically found in found in association with prolonged postprandial hypertrigylceridaemia and low HDL cholesterol, appear more susceptible to oxidation which may make them more atherogenic. Retention of oxidised lipoproteins in the subendothelial space generates an inflammatory reaction – the ‘response to retention’ hypothesis of atherosclerosis
Partially metabolised remnants of triglyceride-rich lipoproteins (remnant lipoproteins) that appear post-prandially are able to induce foam cell formation without modification. These are considered the most highly atherogenic of all. Other atherogenic lipoproteins readily retained in the subendothelial space include glycated LDL and lipoprotein(a). HDL are, however, able to penetrate deep into the subendothelial space and are able to remove oxidised lipid from macrophages and prevent foam cell formation, in addition to having a protective effect on the endothelium. Reduction of HDL particle numbers or functional activity is therefore pro-atherogenic.
Figure 7 shows the progression of atherosclerosis. For more information on the process of atherosclerotic plaque development, please visit module 3 of our angina e-learning programme.
Apolipoproteins
Apolipoproteins are proteins that bind lipids to form lipoproteins. They transport lipids through the lymphatic and circulatory systems. They also serve as enzyme cofactors, receptor ligands, and lipid transfer carriers that regulate the metabolism of lipoproteins and their uptake in tissues. For examples of their beneficial role e.g. apoE in hepatic clearance of chylomicrons and their thrombogenic potential e.g. lipoprotein(a), see dropdown box.
Apolipoproteins
Apolipoproteins B100 and B48 are two proteins produced from the same gene, due to editing of mRNA in the gut. ApoB48 is exclusive to chylomicrons and chylomicron remnants. ApoB48 contains 48% of the sequence of apoB100, and lacks the ligand binding domain recognised by the LDL-receptor (LDLR), so that hepatic removal chylomicron remnants is dependent on ApoE.
ApoE is required for hepatic clearance of chylomicron and VLDL remnants (IDL) via the LDL receptor (LDLR) and LDLR-like receptor protein 1 (LRP1). ApoE has 3 common Isoforms (E3, E4, E2). Of the 6 common genotypes, E3/E3 is the most prevalent. E2 exhibits defective binding to LDLR (<2% of E3) predisposing to remnant (Type III) hyperlipidaemia (familial dysbetalipoproteinaemia), which is usually associated with the E2/E2 genotype.
ApoB100 remains with VLDL as it undergoes lipolysis to IDL and LDL whereas exchangeable apolipoproteins (e.g. ApoE and apoC-II) can transfer between particles. As each LDL particle contains one molecule of apolipoprotein B100, apolipoprotein B concentration is a measure of LDL particle numbers.
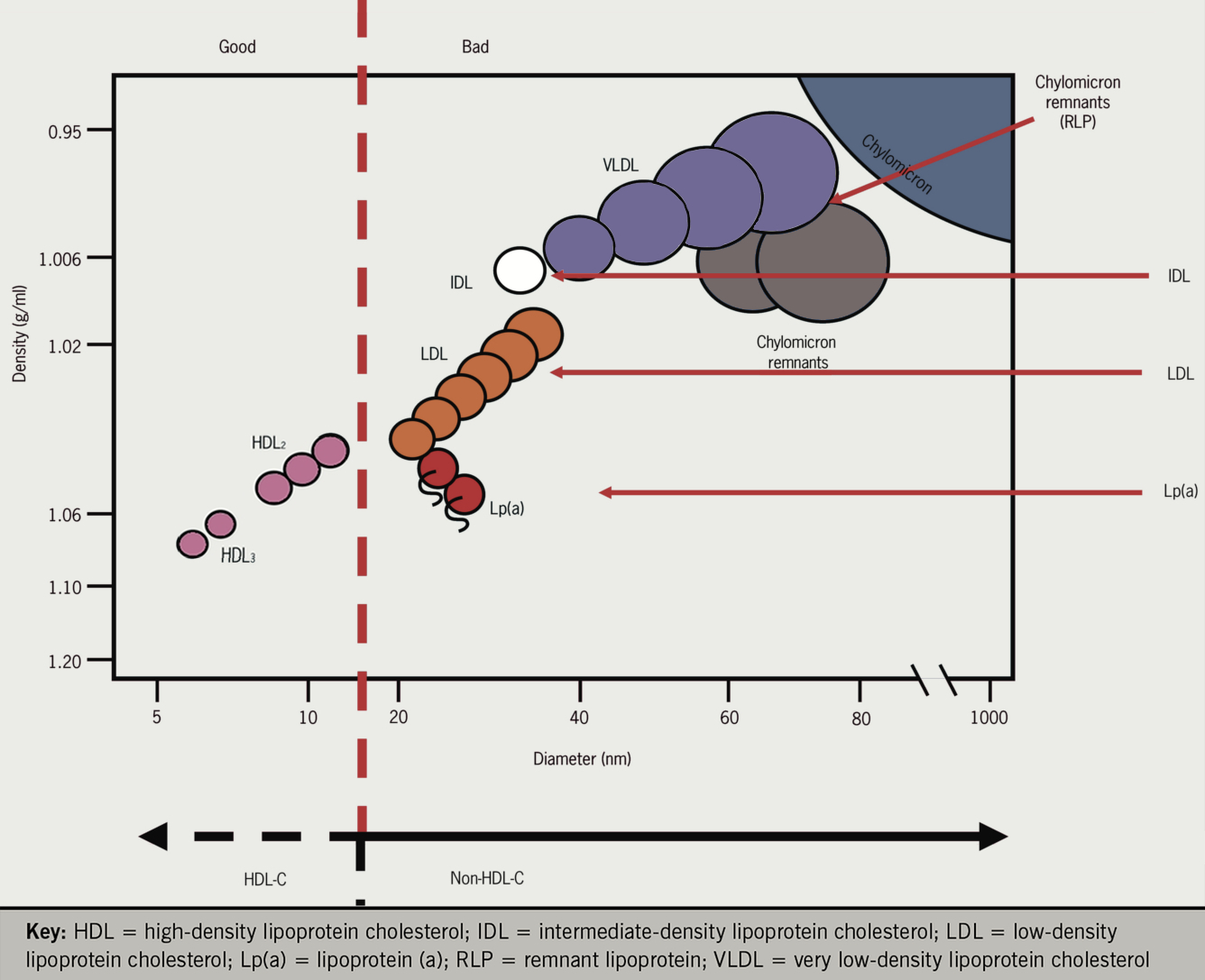
Lipoprotein(a) is a highly atherogenic and thrombogenic lipoprotein formed by covalent bonding between the apolipoprotein B of the LDL particle and apolipoprotein(a), an apparently vestigial plasminogen-like protein which increases its retention in the artery wall (see figure 8). Genetic variants of the apo(a) protein which are smaller in size (due to fewer numbers of repeats of the KIV-2 domain) generate greater numbers of liporotein(a) particles, concentrations of which show 100-fold variation between individuals. As these genetic variants are co-dominantly inherited, family members may also be at risk.1
Routine lipid profile measurements
The routine lipid profile is based on measurements of:
– total cholesterol (TC),
– HDL
– triglycerides
These can be measured easily on automated laboratory analysers and some point of care systems.
 Measuring TC provides limited information about risk because the number includes both atherogenic (LDL, IDL and VLDL) and, the anti-atherogenic fraction, HDL. HDL is essential for calculation of the TC/HDL ratio as is required for cardiovascular risk assessment.
Measuring TC provides limited information about risk because the number includes both atherogenic (LDL, IDL and VLDL) and, the anti-atherogenic fraction, HDL. HDL is essential for calculation of the TC/HDL ratio as is required for cardiovascular risk assessment.
Triglycerides, mainly carried in VLDL, are not considered directly atherogenic but are a risk modifier, a component of the ‘Metabolic syndrome’, a sentinel marker of secondary hyperlipidaemias and a risk factor for pancreatitis when severely elevated (>20 mmol/L). In addition, 12 hour fasting measurement of triglycerides is required for calculation of LDL. LDL is usually considered the most important class of atherogenic lipoproteins. Measurement of LDL is complex so it is usually calculated using the Friedewald equation:
LDL = TC – (HDL + TG/2.2)
However this equation assumes a constant ratio (1:2.2 or 0.45) of cholesterol to triglyceride in VLDL, and requires a fasting sample to ensure absence of postprandial lipoproteins, including chylomicrons and chylomicron remnants and is valid only if fasting triglyceride is less than 4.5 mmol/L. As the ratio is altered by statin treatment, the equation significantly underestimates LDL in treated patients.
Non-HDL is a simple alternative to LDL which is easier to calculate and is preferred when TGs are elevated or the patient has not been fasting when sampled, giving the concentration of total atherogenic lipoproteins:
Non-HDL = TC – HDL
Both LDL and non-HDL measurements are used for targeting treatment but the latter is more reliable in patients treated receiving high intensity statin therapy, since the LDL calculation is increasingly inaccurate at lower concentrations (see table 3).
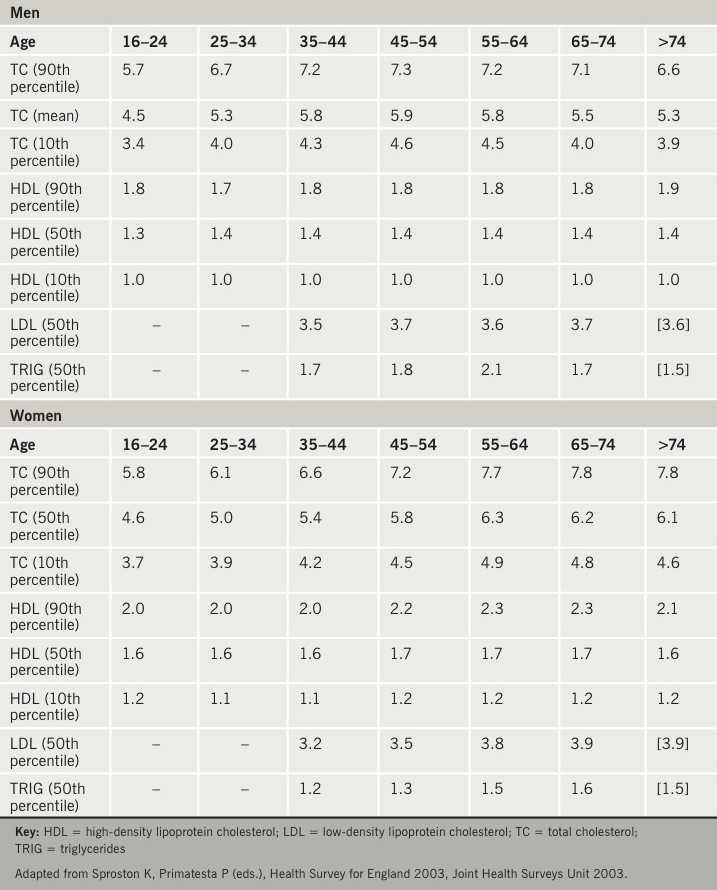
Understanding clinical results
A patient with low LDL and high non-HDL is an example of a patient with increased risk who may slip through the net because we only look at LDL. These patients are also likely to have high LDL particle number (LDL-P) as well as high ApoB levels.
Apolipoproteins are measured by immunoassay and are therefore more expensive tests. ApoB is an alternative measure of atherogenic lipoproteins. The ratio of ApoB/ApoA1, which has been used in epidemiological studies such as INTERHEART,2 provides similar information to the TC/HDL ratio.
In patients with mixed hyperlipidaemia, the ratio of non-HDL (total atherogenic cholesterol) to ApoB is a measure of the average size of atherogenic particles and is characteristically very high (<5 mmol/G) in remnant (type III) hyperlipidaemia (familial dysbetalipoproteinaemia).3
Lipoprotein(a) is also measured by immunoassay and careful method selection is required to ensure equal recognition of particles containing different apo(a) isoforms, but the measurement is stable throughout life and a single measurement is usually sufficient to assess lipoprotein(a) associated cardiovascular risk.1
Therefore there is a need for a lipid parameter that better reflects the amount of cholesterol within all atherogenic particles. This is of particular importance when triglyceride levels are high which is quite common, for example among people with abdominal obesity and/or metabolic syndrome.
Such people have elevated triglycerides, low HDL and relatively normal calculated LDL. Despite this, they harbour highly atherogenic lipoproteins such as TRL remnants and IDL (intermediate density lipoprotein) as well as small dense LDL particles.
Non-HDL has been shown to be a better marker of risk in both primary and secondary prevention studies. In a recent analysis of data combined from 68 studies, non-HDL was the best predictor among all cholesterol measures, both for CAD events and for strokes, and is recommended in the National Institute for Health and Care Excellence (NICE) Lipid Modification Guideline CG181 as the preferred marker for monitoring of lipid lowering therapy.4
close window and return to take test
References
1. Cegla J, Neely RDG, France M, et al.; HEART UK Medical, Scientific and Research Committee. HEART UK consensus statement on lipoprotein(a): a call to action. Atherosclerosis 2019;291:62–70. https://doi.org/10.1016/j.atherosclerosis.2019.10.011
2. Yusuf S, Hawken S, Ounpuu S, on behalf of the INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet 2004;364:937–52. http://dx.doi.org/10.1016/S0140-6736(04)17018-9
3. Boot CS, Middling E, Allen J, Neely RDG. Evaluation of the non-HDL cholesterol to apolipoprotein B ratio as a screening test for dysbetalipoproteinemia. Clin Chem 2019;65:313–20. https://doi.org/10.1373/clinchem.2018.292425
4. National Institute for Health and Care Excellence. Cardiovascular disease: risk assessment and reduction, including lipid modification. Clinical guideline [CG181]. London: NICE, July 2014, updated September 2016. https://www.nice.org.uk/guidance/CG181 (last accessed 5th November 2020)
Background reading
Durrington PN. Hyperlipidaemia: diagnosis and management (3rd Edition). Hodder Arnold 2007. https://doi.org/10.1201/b13464
Garg A (ed). Dyslipidaemias: pathophysiology, evaluation and management. Humana Press 2015. https://doi.org/10.1007/978-1-60761-424-1
Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation 2007;116:1832–44. https://doi.org/10.1161/CIRCULATIONAHA.106.676890
Calandra S, Tarugi P, Speedy HE, Dean AF, Bertolini S, Shoulders CC. Mechanisms and genetic determinants regulating sterol absorption, circulating LDL levels, and sterol elimination: implications for classification and disease risk. J Lipid Res 2011;52:1885–926. https://doi.org/10.1194/jlr.R017855
All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.