Introduction
Table 1. Main amyloid types and responsible precursor fibrils
| Amyloid type | Precursor fibril protein |
|---|---|
| Immunoglobulin light chain (AL) | Immunoglobulin light chains |
| Hereditary transthyretin (hATTR) | Mutations of the transythretin molecules |
| Wild-type transthyretin (wtATTR) | Wild-type (non-mutant) transthyretin molecules |
| Hereditary fibrinogen (AFib) | Mutations of the fibrinogen molecules |
| Hereditary apolipoprotein (AApoA1) | Mutations of apolipoprotein A1 molecules |
| Isolated atrial amyloid (IAA) | Atrial natriuretic peptide |
| Systemic AA | Serum amyloid A (SAA) protein |
| This table illustrates the most common amyloid types and is not inclusive of all types. In TTR amyloidosis the nomenclature is ATTR preceded by ‘wt’ to indicate the wild-type protein or ‘h’ to indicate a hereditary variant of transthyretin. | |
The amyloidoses are a rare group of diseases that result from the extracellular distribution of amyloid, a fibrillar material derived from a range of precursor fibrils that aggregate with a highly abnormal cross-beta-sheet conformation.1,2 These deposits progressively disrupt the structure of tissues and affect organ function.3 Amyloid is identified by the pathognomonic finding of apple-green birefringence when tissue is stained with the dye Congo red and visualised under cross-polarised light. Amyloid type is classified according to the fibril protein, with over 30 fibril proteins identified in vivo (table 1).4 Amyloidosis is a heterogeneous condition in which deposition can range from incidental localised deposits, in a range of organs, to a rapid and fatal systemic disease. Cardiac amyloidosis, the most feared manifestation, is associated with significant morbidity and mortality and typically exhibits; a restrictive cardiomyopathy; concentric ventricular wall thickening resulting in diastolic and longitudinal systolic dysfunction manifesting with congestive cardiac failure and frequent hypotension.
Hereditary amyloidosis, which has a worldwide distribution, is most commonly caused by genetic variants of the transthyretin (TTR) protein and manifests primarily with cardiomyopathy (hATTR-CM), polyneuropathy (hATTR-PN) or a combination of both (hATTR-mixed).
Wild-type transthyretin (wtATTR) cardiac amyloidosis
Wild-type transthyretin amyloidosis (wtATTR) previously termed ‘senile systemic/cardiac amyloidosis’ is a disease of older people with a strong male preponderance. Amyloid deposits are composed of wild-type TTR,5 and autopsy series have reported the presence of wtATTR amyloid in 25% of patients over 80 years of age.6 It is important to note that presence of wtATTR deposits histologically does not prove the existence of wtATTR amyloid cardiomyopathy (ATTR-CM). However, the clinical phenotype of wtATTR amyloidosis is dominated by cardiac amyloidosis manifesting as congestive cardiac failure, despite deposits reported in other sites including the lungs, gut and bladder, where they occasionally cause symptoms.7 Soft-tissue ATTR amyloid deposits manifesting as carpal tunnel syndrome and spinal canal stenosis are common, and often precede cardiac manifestations by up to a decade or more.8
Hereditary transthyretin cardiac amyloidosis (hATTR-CM)
hATTR amyloidosis has historically been split into two phenotypes, those with a predominant neurological presentation, known as hATTR-PN, and those with a cardiac presentation, hATTR-CM, with a significant overlap between the two classifications (hATTR-mixed). In both forms of hATTR amyloidosis, a family history is reported in just under half of patients,9 and there are over 130 different TTR mutations recognised.10 Age-related effect on phenotype is seen in patients with the common TTR variant Val30Met, with neuropathy predominating in early onset and cardiomyopathy in late-onset disease.11,12 hATTR occurs worldwide and while Val30Met was first identified in Portugal, it is also endemic in Sweden, Brazil and Japan, and is the most commonly reported mutation to the Familial Amyloid Polyneuropathy World Transplant Registry (FAPWTR).13 The TTR Val122Ile variant is also common and is carried by almost 4% of African-American and Afro-Caribbean populations, with an uncertain disease penetrance, but is a recognised risk factor for the development of ATTR-CM.14,15
Management of ATTR cardiac amyloidosis
There are four key principles in the management of ATTR amyloidosis. Supportive care to preserve organ function, reduction and ideally elimination of TTR from the plasma, stabilisation of the tetramic structure of TTR and dissolution of the existing ATTR amyloid matrix. A further potential therapeutic target includes reduction of the ‘toxic’ oligomeric precursors.
Supportive care
The supportive care of ATTR cardiac amyloidosis requires a comprehensive approach including treatment of heart failure symptoms, management of arrhythmias and other conduction abnormalities, systemic anticoagulation when appropriate and management of extra-cardiac manifestations.
Heart failure therapy
Optimisation of heart failure (HF) symptoms in cardiac amyloidosis is challenging as hypotension is poorly tolerated due to the low cardiac output state. Current management focuses on meticulous fluid balance control through dietary counselling to encourage sodium restriction and the use of diuretics. A combination of loop diuretics with a mineralocorticoid receptor antagonist (MRA) is often felt to be the most effective approach in clinical practice.16
While current HF guidelines recommend neurohormonal blockade, there remains no evidence of prognostic benefit with traditional HF medications, such as beta blockers, angiotensin-converting enzyme (ACE) inhibitors/angiotensin-receptor blockers (ARBs), and MRAs, in cardiac ATTR amyloidosis, some of which may be poorly tolerated in this patient cohort, often due to exacerbation of hypotension. Results from a single-centre retrospective cohort study demonstrated reduced survival in patients with ATTR-CM treated with ACE inhibitors and beta blockers.17
Atrial arrhythmias
Conduction abnormalities are common in patients with cardiac amyloidosis,18 and include both brady- and tachyarrythmias. In a retrospective cohort of over 260 patients with ATTR-CM, atrial fibrillation (AF) was seen in 14.5% of patients and was more common in older patients and those with wild-type disease.19 Despite no prognostic benefit of beta blockers, they have a role as rate-controlling agents. Nondihydropyridine calcium channel blockers, such as verapamil and diltiazem, tend to be avoided since they bind to amyloid fibrils and may induce syncope from severe hypotension.16 Digoxin is used with caution due to concerns about increased risk of toxicity and sudden death,20 although it is not contraindicated in patients with challenging rate control. Anticoagulation is recommended in all patients with AF and cardiac amyloidosis irrespective of CHAD-VASC score as there is a high incidence of left atrial thrombus in this patient population. There are some data to suggest consideration of systemic anticoagulation in patients with enlarged atria, even when in sinus rhythm.21
Pacemakers and implantable cardiac defibrillators
There are limited data on the role of implantable intracardiac defibrillators (ICD) in patients with cardiac amyloidosis. While appropriate device therapy has been reported in a significant proportion of patients with light-chain cardiac amyloidosis, there remains no strong evidence of long-term survival benefit,22 which may be due to the importance of electromechanical dissociation (EMD) as a cause of sudden death in this population.23 Implantation of pacemakers for life-threatening bradyarrhythmias, or symptoms such as syncope, should follow current standard guidelines, and European guidelines recommend an ICD be considered in patients with ventricular arrhythmias causing haemodynamic instability, who have a good functional status and a prognosis of greater than one year.24
Extra-cardiac manifestations
Amyloid-related autonomic nerve dysfunction is predominantly seen in hATTR amyloidosis, but can be a feature of wtATTR, and predominantly manifests with postural hypotension, weight loss, altered bowel habit, incontinence and erectile dysfunction. Anecdotal evidence supports the use of oral inotropes, such as midodrine, for the treatment of postural hypotension. While symptoms due to direct gastrointestinal (GI) amyloid infiltration are thought to be rare, presence of ATTR amyloid deposits in the GI tract is commonly reported. GI symptoms such as chronic diarrhoea, malabsorption and cachexia, usually represent autonomic disturbance and often becoming debilitating. Somatostatin analogues have provided relief in some case studies, but if symptoms lead to malnutrition, total parenteral nutrition may be required.
Disease-modifying therapy
Orthotropic liver transplantation
When the liver is the sole source of circulating variant amyloidogenic precursor protein, liver transplantation (LT) can be performed to replace the variant protein with the wild-type ‘non-amyloidogenic’ protein, the rationale being elimination of the circulating amyloid fibril precursor. This approach has been employed in a variety of hereditary amyloidoses, most commonly hATTR amyloidosis. LT for hATTR amyloidosis began in 1991, with international, 10-year experience, demonstrating an overall 5-year survival of 77%, comparable to LT performed for other liver disorders.25 By the end of 2012, over 2,060 liver transplants had been recorded on the FAPWTR. However, while circulating variant TTR concentration is reduced by >95% following LT, it became apparent that patients suffered with progression of both their cardiac, and less so, neurological symptoms.26 This was later determined to be due to the deposition of wild-type ATTR amyloid, produced by the donor liver, on a template of variant ATTR amyloid deposits with a specific predilection for the myocardium. In light of this, careful consideration is now given to selection of patients for LT, with FAPWTR data indicating a 10-year survival of 74% in Val30Met compared with 44% in non-Val30Met patients: a Japanese cohort of 32 patients with early onset Val30Met variant demonstrated 100% survival at 10 years.27 While LT remains the therapy that has unequivocally been shown to improve survival in carefully selected patients, its use now tends to be restricted to younger Val30Met patients (<50 years) with a short disease duration (<7 years) and no cardiac amyloidosis.28-30 Further consideration needs to be given to the fact that LT is a major surgical procedure with a requirement for long-term immunosuppression and follow-up.
RNA-targeted therapies
RNA-targeted therapies aimed at inhibiting the production of TTR protein by the liver have demonstrated great promise in recent clinical trials and include antisense oligonucleotides (ASOs)31 and small interfering RNAs (siRNAs).32 Both patisiran (siRNA) and inotersen (ASO) have recently been licensed and approved by the National Institute for Health and Care Excellence (NICE) for the treatment of hereditary transthyretin amyloidosis with evidence of polyneuropathy. While there are differences in the chemical structures and delivery methods of the two classes of drugs, they have key similarities including:
- Binding to the 3’ untranslated region (3’UTR) of TTR mRNA.
- Harnessing intracellular mechanisms that result in the degradation of TTR mRNA.
- Reduction of both wild-type and variant circulating liver-derived TTR concentration.
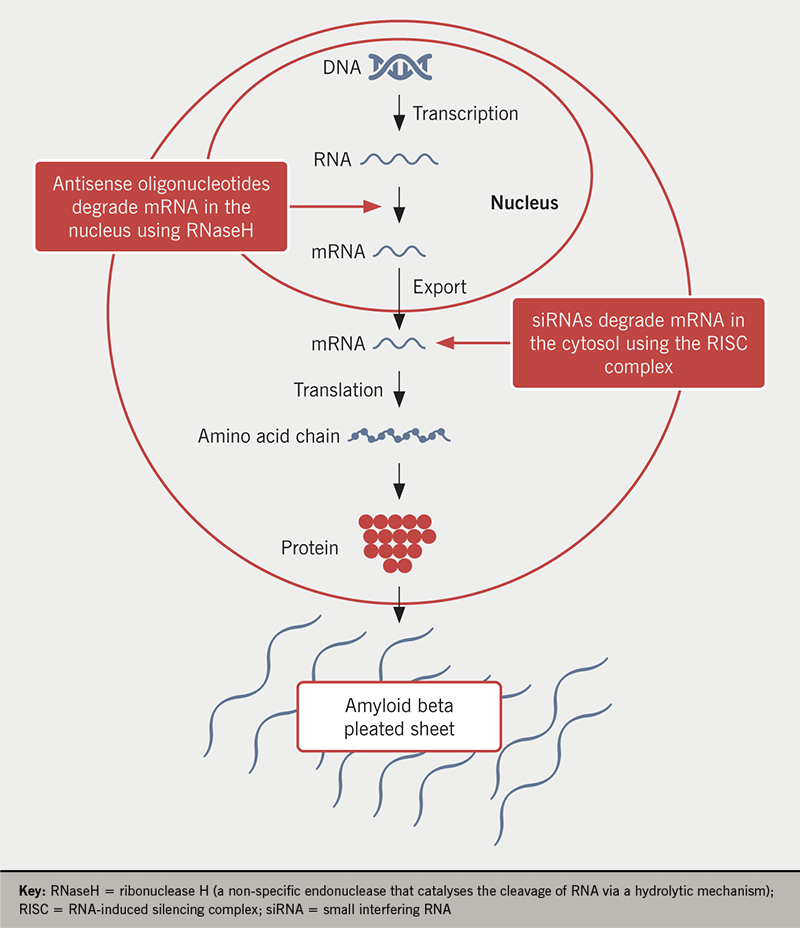
siRNAs
siRNAs are double-stranded RNAs that bind to the RNA-induced silencing complex and mediate cleavage of the target mRNA (figure 1). There are currently two therapies in clinical use; patisiran (ALN TTR02), which is administered by intravenous infusion every three weeks, and vutrisiran (ALN TTRSC02), which is administered subcutaneously every three months. Both agents result in median serum TTR knockdown of 80–85%.
APOLLO, a phase 3 randomised, double-blind, placebo-controlled trial, in patients with hereditary ATTR amyloidosis and polyneuropathy, demonstrated that patisiran improved multiple clinical manifestations including; Norfolk Quality of Life–Diabetic Neuropathy (Norfolk QOL–DN) score, modified Neuropathy Impairment Score+7 (mNIS+7), gait speed and modified body mass index (BMI).33 Subpopulation analysis also demonstrated improvement in key cardiac parameters including reduction in; left ventricular wall thickness, global longitudinal strain, N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels, as well as adverse cardiac outcomes compared with placebo at month 18, suggesting that patisiran may halt or reverse progression of ATTR-CM.34 In light of this, a second study, APOLLO-B is currently underway to evaluate patisiran in patients with ATTR-CM (both hereditary and wild-type).
HELIOS-A, a phase 3 global, randomised, open-label study to evaluate the efficacy and safety of vutrisiran in patients with hATTR-PN, and HELIOS-B evaluating its effect in patients with ATTR-CM (both hereditary and wild-type) are in progress.
ASOs
Inotersen (ISIS-TTRRx) is a second-generation ASO drug designed to target human TTR mRNA. The hybridisation (binding) of inotersen to the mRNA results in the RNaseH-mediated degradation of the TTR mRNA, thus reducing production of the TTR protein (figure 1). As inotersen binds to a non-mutation site, it results in the reduction of both wild-type and variant forms of TTR produced by the liver. NEURO-TTR, a global, randomised, double-blind, placebo-controlled phase 3 study in 172 patients with hATTR-PN demonstrated statistically significant improvement in the primary end points of Norfolk QOL–DN score and mNIS+7 at week 66.35 Importantly, both ASOs and siRNAs are not predicted to reduce levels of TTR in the central nervous system (CNS) because of their inability to cross the blood–brain barrier. This may be of clinical significance since TTR has been reported to have beneficial neuroprotective effects, by binding to beta-amyloid protein in the brain and inhibiting accumulation in Alzheimer’s disease plaques,36 but may also lead to a change in the future phenotype of hATTR amyloidosis, with complications from ongoing leptomeningeal amyloid deposition.
TTR stabilisation
There are several agents that have been demonstrated to stabilise the tetramic structure of TTR and prevent its dissociation into its kinetically unstable monomer units.37 The three main drugs known to do this are tafamidis, which is an approved therapy for the treatment of hATTR-PN and ATTR-CM, acoramidis (AG10) and diflunisal.
Tafamidis meglumine
In vitro studies have shown that amyloidogenic misfolding of TTR may be inhibited by compounds that bind TTR in the plasma. Tafamidis was the first disease-modifying drug to be approved for hATTR-PN and was shown to slow neuropathic disease in patients with Val30Met disease.38 It binds to the thyroxine-binding site of the tetrameric structure of TTR to decrease dissociation into monomeric units. It has recently been tested in a multi-centre, international, double-blind, placebo-controlled, phase 3 trial (ATTR-ACT), of over 440 patients with ATTR-CM, both variant and wild-type, who received 80 mg of tafamidis, 20 mg of tafamidis, or placebo for 30 months. Tafamidis was associated with a reduction in all-cause mortality and cardiovascular-related hospitalisations compared with placebo, and slowed the decline in both functional capacity and quality of life.39
Acoramidis (AG10)
Dissociation or selective mechanoenzymatic cleavage of the native TTR tetramer is an essential step in ATTR fibrillogenesis, and pathogenic TTR variants are known to destabilise the native tetramer. Furthermore, a naturally occurring super-stabilising mutation (T119M) has been identified that protects pathogenic TTR mutation carriers from development of hATTR amyloidosis by reducing the dissociation rate of the tetramic TTR by 30-fold compared with its wild-type counterpart.40 Acoramidis is a potent, highly selective TTR stabiliser that was designed to mimic the structural influence on TTR of the protective T119M variant. A phase 2 randomised, double-blind, placebo-controlled study in 49 patients with ATTR-CM reported that acoramidis was well tolerated, achieved target plasma concentrations and demonstrated near complete in vitro stabilisation of TTR. It, therefore, has the potential to be a safe and effective treatment for ATTR-CM with a phase 3 trial currently ongoing.41
Diflunisal
Diflunisal is a non-steroidal anti-inflammatory drug that binds to and stabilises TTR in vitro, and has been repurposed as an amyloid treatment.42 A randomised, placebo-controlled trial showed a slowing of neurological progression in hATTR amyloidosis and a slowing of the decline in quality of life,43 although it has not been approved for this indication. Due to its potent non-steroidal anti-inflammatory effects, caution is advised with its use in patients with impaired renal function and prior GI bleeding.
Conclusion
The diagnosis and management of patients with ATTR amyloidosis is challenging, with many patients waiting a number of years before diagnosis. Therapy is aimed at supportive care of the failing amyloidotic organ, with important considerations including the specific disease pathophysiology and its frequent multi-system nature, and a lack of evidence for use of certain medications. Disease-modifying therapy is focused upon either reduction of circulating TTR concentration or stabilisation of the TTR tetramer while ‘surgical gene therapy’ in the form of liver transplantation remains a therapeutic option in selected patients with hATTR-PN who do not have pre-existing amyloid cardiomyopathy. It is likely that future management of cardiac ATTR amyloidosis, both variant and wild-type will focus on promising RNA-targeting therapies and TTR stabilisers. The rapid evolution of treatment options for ATTR cardiac amyloidosis justifies more extensive efforts to obtain a diagnosis, particularly of wild-type ATTR amyloidosis among elderly patients with heart failure.
Key messages
- Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and fatal condition
- Current management focuses on supportive care, namely with meticulous control of fluid status, anti-arrhythmic therapy and systemic anticoagulation
- Novel disease-modifying therapies are aimed at:
- Reduction and ideally elimination of TTR from the plasma – antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) therapies
- Stabilisation of the tetramic structure of TTR (tafamidis and acoramidis)
Conflicts of interest
TR: None declared. JG is an expert advisor for Alnyam Pharamaceuticals Inc., Ionis/Akcea Therapeutics and Eidos Therapeutics Inc.
close window and return to take test
References
1. Lachmann HJ, Hawkins PN. Systemic amyloidosis. Curr Opin Pharmacol 2006;6:214–20. https://doi.org/10.1016/j.coph.2005.10.005
2. Merlini G. Systemic amyloidosis: are we moving ahead? Neth J Med 2004;62:104–05. https://doi.org/10.1182/blood-2004-06-2451
3. Pepys MB. Amyloidosis. In: Frank MM, Austen KF, Claman HN, Unanue ER, eds. Samter’s Immunologic Diseases. Fifth ed. Boston: Little, Brown and Company, 1994;pp. 637–55.
4. Sipe JD, Benson MD, Buxbaum JN et al. Nomenclature 2014: amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014;21:221–4. https://doi.org/10.3109/13506129.2014.964858
5. Westermark P, Sletten K, Johansson B, Cornwell GG. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA 1990;87:2843–5. https://doi.org/10.1073/pnas.87.7.2843
6. Cornwell GG 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkanen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med 1983;75:618–23. https://doi.org/10.1016/0002-9343(83)90443-6
7. Westermark P, Bergstrom J, Solomon A, Murphy C, Sletten K. Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid 2003;10(suppl 1):48–54. https://doi.org/10.1080/13506129.2003.12088568
8. Youngstein T, Gilbertson JA, Hutt DF et al. Carpal tunnel biopsy identifying transthyretin amyloidosis. Arthritis Rheumatol 2017;69:2051. https://doi.org/10.1002/art.40162
9. Swiecicki PL, Zhen DB, Mauermann ML et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid 2015;22:123–31. https://doi.org/10.3109/13506129.2015.1019610
10. Rowczenio DM, Noor I, Gillmore JD et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat 2014;35:E2403–E2412. https://doi.org/10.1002/humu.22619
11. Hattori T, Takei Y, Koyama J, Nakazato M, Ikeda S. Clinical and pathological studies of cardiac amyloidosis in transthyretin type familial amyloid polyneuropathy. Amyloid 2003;10:229–39. https://doi.org/10.3109/13506120309041740
12. Hornsten R, Pennlert J, Wiklund U, Lindqvist P, Jensen SM, Suhr OB. Heart complications in familial transthyretin amyloidosis: impact of age and gender. Amyloid 2010;17:63–8. https://doi.org/10.3109/13506129.2010.483114
13. Wilczek HE, Larsson M, Ericzon BG. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid 2011;18(suppl 1):193–5. https://doi.org/10.3109/13506129.2011.574354072
14. Quarta CC, Buxbaum JN, Shah AM et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med 2015;372:21–9. https://doi.org/10.1056/NEJMoa1404852
15. Gillmore JD, Hawkins PN. V122I transthyretin variant in elderly black Americans. N Engl J Med 2015;372:1769. https://doi.org/10.1056/NEJMc1503222
16. Ritts AJ, Cornell RF, Swiger K, Singh J, Goodman S, Lenihan DJ. Current concepts of cardiac amyloidosis: diagnosis, clinical management, and the need for collaboration. Heart Fail Clin 2017;13:409–16. https://doi.org/10.1016/j.hfc.2016.12.003
17. Aus dem Siepen F, Hein S, Bauer R, Katus HA, Kristen AV. Standard heart failure medication in cardiac transthyretin amyloidosis: useful or harmful? Amyloid 2017;24(suppl 1):132–3. https://doi.org/10.1080/13506129.2016.1272453
18. Pinney JH, Whelan CJ, Petrie A et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013;2:e000098. https://doi.org/10.1161/JAHA.113.000098
19. Longhi S, Quarta CC, Milandri A et al. Atrial fibrillation in amyloidotic cardiomyopathy: prevalence, incidence, risk factors and prognostic role. Amyloid 2015;22:147–55. https://doi.org/10.3109/13506129.2015.1028616
20. Castano A, Drachman BM, Judge D, Maurer MS. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev 2015;20:163–78. https://doi.org/10.1007/s10741-014-9462-7
21. Dubrey S, Pollak A, Skinner M, Falk RH. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Br Heart J 1995;74:541–4. https://doi.org/10.1136/hrt.74.5.541
22. Rezk T, Whelan CJ, Lachmann HJ et al. Role of implantable intracardiac defibrillators in patients with cardiac immunoglobulin light chain amyloidosis. Br J Haematol 2018;182:145–8. https://doi.org/10.1111/bjh.14747
23. Yamamoto H, Yokochi T. Transthyretin cardiac amyloidosis: an update on diagnosis and treatment. ESC Heart Fail 2019;6:1128–39. https://doi.org/10.1002/ehf2.12518
24. Priori SG, Blomstrom-Lundqvist C, Mazzanti A et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J 2015;36:2793–867. https://doi.org/10.1093/eurheartj/ehv316
25. Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation 2004;77:64–71. https://doi.org/10.1097/01.TP.0000092307.98347.CB
26. Stangou AJ, Hawkins PN, Heaton ND et al. Progressive cardiac amyloidosis following liver transplantation for familial amyloid polyneuropathy: implications for amyloid fibrillogenesis. Transplantation 1998;66:229–33. https://doi.org/10.1097/00007890-199807270-00016
27. Yamashita T, Ando Y, Okamoto S et al. Effect of liver transplantation on the survival of patients with ordinary onset familial amyloid polyneuropathy in Japan. Amyloid 2011;18(suppl 1):185–6. https://doi.org/10.3109/13506129.2011.574354069
28. Stangou AJ, Hawkins PN. Liver transplantation in transthyretin-related familial amyloid polyneuropathy. Curr Opin Neurol 2004;17:615–20. https://doi.org/10.1097/00019052-200410000-00012
29. Okamoto S, Wixner J, Obayashi K et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients’ survival. Liver Transpl 2009;15:1229–35. https://doi.org/10.1002/lt.21817
30. Ericzon BG, Wilczek HE, Larsson M et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation 2015;99:1847–54. https://doi.org/10.1097/TP.0000000000000574
31. Benson MD, Kluve-Beckerman B, Zeldenrust SR et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve 2006;33:609–18. https://doi.org/10.1002/mus.20503
32. Coelho T, Adams D, Silva A et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369:819–29. https://doi.org/10.1056/NEJMoa1208760
33. Adams D, Gonzalez-Duarte A, O’Riordan WD et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018;379:11–21. https://doi.org/10.1056/NEJMoa1716153
34. Solomon SD, Adams D, Kristen A et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019;139:431–43. https://doi.org/10.1161/CIRCULATIONAHA.118.035831
35. Brannagan TH, Wang AK, Coelho T et al. Early data on long-term efficacy and safety of inotersen in patients with hereditary transthyretin amyloidosis: a 2-year update from the open-label extension of the NEURO-TTR trial. Eur J Neurol 2020;27:1374–81. https://doi.org/10.1111/ene.14285
36. Schwarzman AL, Gregori L, Vitek MP et al. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci USA 1994;91:8368–72. https://doi.org/10.1073/pnas.91.18.8368
37. Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science 2003;299:713–16. https://doi.org/10.1126/science.1079589
38. Coelho T, Maia LF, Martins da Silva A et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79:785–92. https://doi.org/10.1212/WNL.0b013e3182661eb1
39. Maurer MS, Schwartz JH, Gundapaneni B et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16. https://doi.org/10.1056/NEJMoa1805689
40. Hesse A, Altland K, Linke RP et al. Cardiac amyloidosis: a review and report of a new transthyretin (prealbumin) variant. Br Heart J 1993;70:111–15. https://doi.org/10.1136/hrt.70.2.111
41. Judge DP, Heitner SB, Falk RH et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol 2019;74:285–95. https://doi.org/10.1016/j.jacc.2019.03.012
42. Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236–49. https://doi.org/10.1080/13506120600960882
43. Berk JL, Suhr OB, Obici L et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA 2013;310:2658–67. https://doi.org/10.1001/jama.2013.283815

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.