Patients with amyloid heart disease have historically been considered to have a very poor prognosis and were considered almost untreatable. However, recent therapeutic advances are encouraging and likely to have a marked effect on management across the amyloid spectrum. This message needs to be conveyed to cardiologists, not least because there is now benefit to performing an endomyocardial biopsy to determine amyloid type. We provide an update on the significant progress in managing the three most common forms of amyloid heart disease in the UK.
Introduction
The amyloidoses comprise a variety of proteins that are deposited in various organ systems. The major categories of amyloid seen in the UK are shown in figure 1,1 although low detection rates for some types (e.g. senile systemic amyloid) may mean the breakdown in figure 1 does not reflect the true prevalence and proportions of each amyloid type.
Without therapy, the consequences of significant heart involvement are inevitably fatal. The main variable on life-expectancy being amyloid type, extent of systemic and, particularly, heart involvement. Historically considered untreatable, this view of amyloidosis remains embedded, particularly in the mind of the cardiologist.
As with almost every cause of severe cardiomyopathy, treatments may be limited by the comorbidities of haemodynamic instability, renal, hepatic and pulmonary involvement. While the treatment of amyloid related heart failure remains fairly static,2 considerable progress has been achieved in treating the underlying amyloidogenic process. In brief, heart failure is managed with carefully monitored use of diuretics. Combinations of loop and potassium-sparing diuretics (spironolactone or epleronone) may have an effective synergistic effect, while carefully monitoring a risk of hypotension. Angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, beta blockers and digoxin are poorly tolerated. Hypotension may necessitate compression stockings and the vasopressor alpha-agonist midodrine. There remains little evidence of benefit, in terms of survival, from the use of amiodarone or implantable defibrillators.3,4 Mechanical dysfunction of the atria due to amyloid infiltration and often severe dilatation, either with or without atrial fibrillation, means anticoagulation with warfarin should be seriously considered. The general management of heart failure due to amyloid involvement of the heart is extensively reviewed elsewhere.5,6
Immunoglobulin light-chain AL amyloidosis
Amyloid light-chain (AL) amyloidosis is caused by the deposition and assembly of immunoglobulin light chains into a fibrillar amyloid matrix. Treatment of the disease process and dissolution of amyloid is now feasible.7 However, when the heart is involved, several months may elapse following therapy before measureable benefit is evident.
Introduction to the treatment of AL amyloidosis
Disease-modifying therapies in AL amyloidosis continue to follow advances seen in the field of multiple myeloma. AL amyloidosis is frequently a rapidly fatal disease. Crucial to management is a therapy that will quickly remove amyloidogenic precursor proteins. This produces immediate benefit as component immunoglobulin light chains are inherently cytotoxic. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B are shown to decrease simultaneously in association with improvement of survival in AL amyloidosis.8 Regardless of the type of chemotherapy used, a reduction in amyloidogenic free light chains (FLC) of more than 50% results in a substantial survival benefit.9
High-dose chemotherapy and autologous stem cell transplantation
High rates of haematological, and organ response, have now been achieved by multiple centres. Median survivals of over a decade, for high-dose melphalan (HDM) and autologous stem cell transplantation (ASCT), are reported in patients achieving complete haematological response (CR).10-12 The Boston University group reported on 312 patients and described a median survival, in those achieving a CR, in excess of 10 years, compared with 50 months in those who did not.13 The Mayo Clinic describe 434 patients treated with HDM and ASCT over 14 years, demonstrating a CR in 39%. A median survival was not reached for CR patients but was 107 months in partial responders, compared with 32 months in non-responders.14
Actual regression of cardiac amyloid involvement, has been reported in patients achieving CR undergoing treatment with HDM and ASCT.15,16 The Boston group reported a reduction in left ventricular wall thickness of 1.07 ± 1.98 mm in patients (n=21) with a complete haematologic response to HDM and ASCT. Patients without a complete haematologic response (n=34) showed an increase in wall thickness of 0.37 ± 2.21 mm (p=0.0018).15
In 2007, the obvious success of HDM and ASCT therapy was challenged by the results from a French study. In 100 patients from 29 centres, the haematological results were similar when patients treated with HDM and ASCT were compared with conventional oral melphalan and dexamethasone (67% vs. 68%). Moreover, overall survival was superior in the conventional chemotherapy cohort.17 Several reasons have been proposed for these findings, but are beyond this brief review.18 These findings have resulted in the USA favouring HDM and ASCT, and Europe (with the possible exception of Germany) favouring combination chemotherapy.
New therapies and triple-therapy regimens
Treatment regimens consisting of high-dose steroid (usually dexamethasone) and alkylating agents (usually melphalan or cyclophosphamide and, more recently, bendamustine) are being increasingly challenged (figure 2).
A product that delivers one of the most rapid responses is the reversible proteosome inhibitor bortezomib (Velcade). Kastritis et al. describe a high rate of haematologic responses when bortezomib was prescribed either with or without dexamethasone. A cardiac response, in terms of a sustained improvement in functional class and, in some, a decrease in wall thickness, was seen in 29% of patients.19 A later study, in patients who had relapsed on all prior conventional therapies, suggested bortezomib slowed the progression of cardiac amyloid disease.20 Bortezomib can be used as a stand-alone therapy or within combinations. Patients with Mayo Clinic class III high-risk cardiac amyloidosis have achieved rapid haematological responses to the combination of bortezomib, cyclophosphamide and dexamethasone (VCD or CYBORD regimens).21,22 Moreover, bortezomib alone or with dexamethasone has also proved successful as consolidation therapy following ASCT.23 Second-generation proteosome inhibitors are also being developed, including the irreversible product carfilzomib and the agent MLN9708.
A combination of cyclophosphamide, thalidomide and dexamethasone (CTD) has been demonstrated to deliver a response rate in AL amyloidosis higher than that previously reported for non-transplant (ASCT) regimens and with a lower treatment-related mortality.24
Immuno-modulatory products, initially comprising thalidomide, but lately the derivatives lenalidomide and pomalidomide, are now being used. Among several combinations, a trial in 35 patients (50% with significant cardiac involvement) using lenalidomide with cyclophosphamide and dexamethasone achieved an overall haematological response of 60%.25 Pomalidomide in combination with dexamethasone has also produced very encouraging results, including in previously treated patients (48% had relapsed to prior treatment) and in whom 82% had cardiac involvement.26 Toxicity, particularly peripheral neuropathy and thromboembolic events, remain a considerable issue with thalidomide and its derivatives.
These latest drug combinations have proved so effective that discussion now centres on which to use as first-line therapy.
Novel therapeutic approaches
One approach is to target the ‘chaperone’ glycoprotein, serum amyloid P (SAP), found in abundance in amyloid deposits, and likely crucial to amyloid assembly.27 To date, approaches have used the bis-d-proline compound CPHPC, which binds to circulating SAP and forms complexes that are cleared by the liver. Having depleted the supply of circulating SAP, tissue-bound SAP can then be targeted with anti-SAP monoclonal antibodies.28 A fully humanised version of anti-SAP monoclonal antibody is under investigation.
Heart transplantation and concomitant therapies
Cardiac transplantation, for amyloid heart involvement, has been plagued by recurrence in the graft, as well as with continued systemic deposition. Attempts to prevent this have resulted in many units using modified regimens of melphalan and ASCT following cardiac transplantation (figure 2), with reasonable success. Recent UK data show that of 14 patients receiving heart transplants, the five-year survival was 45%. Eight patients had subsequent ASCT with a median survival of 9.7 years.29 Heart transplantation can also be pre-treated with a ‘risk-adapted’ therapeutic approach, followed-up with consolidation therapy after transplantation, utilising many of the new therapies described above. However, chemotherapeutic consolidation and continuation maintenance therapies have to be balanced against the risk of developing secondary neoplastic disease from these drugs.
Hereditary amyloidosis
Most cases of hereditary amyloidosis (ATTR) are caused by one of over a hundred known mutations of the protein transthyretin (TTR). Without treatment this is a lethal disease, although, with a life-expectancy considerably longer than for AL amyloidosis. Death is usually within 10 years of symptom onset, and frequently due to significant heart involvement.
Transthyretin is a tetrameric protein, produced predominantly from the liver, and responsible for transporting thyroxine and retinal binding protein. Of the 75 mutations currently known to express a clinical phenotype, around 44 (59%) result in cardiomyopathy.30 Of particular interest is a mutation of TTR (Val122Iso) almost exclusively reported in Afro-Caribbeans and with a predilection for the heart. Around 4% of Afro-Caribbeans may carry the genotype.31 A report from the UK indicated that 10% of such patients, attending with cardiomyopathy and heart failure, possessed this Val122Iso mutation.32 Patients with the Val122Iso mutation have a median survival of three to five years, reduced to between 27 and 36 months when cardiomyopathy and heart failure are apparent.32-33 One centre reports a worse survival of 50% at 11 months in patients with this mutation presenting with cardiomyopathy.34
Until very recently, the accepted treatment was orthotopic liver transplantation (OLT) to remove the main source of the mutant protein (figure 3). Over 1,900 OLTs have now been performed in over 70 centres worldwide.35 Initially considered a curative procedure, it is now appreciated that progression of the disease can occur, due to deposition of wild-type transthyretin. Unfortunately, the heart is a particularly avid target for this process.36 Liver transplantation appears a reasonable therapy, if performed early in the disease, in well-nourished patients and in patients possessing the genotype for the most common (worldwide) mutation of TTR (Val30Met).37 On the negative side, liver transplantation is an expensive procedure, requires an organ donor, is not without risk and necessitates life-long administration of immunosupressants.
(TTR) amyloidosis
Transthyretin stabilisers as treatments for hereditary amyloidosis
A major advance in amyloid therapy has been the development of molecules that will stabilise circulating transthyretin, preventing a conformational change that would have resulted in auto-aggregation to form amyloid deposits (figure 3). Around 99% of the thyroxine receptors on TTR molecules are vacant and occupancy of these sites imposes kinetic stability.38 Foremost among these is the orally administered product, tafamidis meglumine (Vindaqel). A study of 128 patients showed a greater proportion of tafamidis-treated patients exhibiting no disease progression, compared with placebo.39 Currently, tafamidis is approved for use in Europe, but is only indicated for the treatment of neuropathic aspects of ATTR.40 A recent trial demonstrated tafamidis to stabilise TTR in most patients with mutant (including the common Val30Met and also non-Val30Met mutations) or wild-type TTR, resulting in less neurological or cardiac deterioration and a maintained quality of life.41
The non-steroidal anti-inflammatory diflunisal has also been demonstrated to prevent dissociation of TTR and fibril formation (figure 3).42 An ongoing trial in ATTR patients is due to report in early 2013, but is currently showing diflunisal to be well tolerated.43 In contrast the UK amyloid centre report a high proportion of adverse events from diflunisal (250 mg twice daily), despite the routine co-prescription of proton-pump inhibitors. The outcome results, of overall efficacy, are awaited.44
Oligosense anti-nucleotide and interfering RNA treatments for TTR amyloidosis
Anti-sense oligonucleotides (ASOs) have been engineered to target both ‘wild’ and all known mutant forms causing ATTR (figure 4). Maximal antisense-mediated reductions of target mRNA levels are typically greater than 90% of control levels, with animal and phase I human studies demonstrating dramatic reductions in circulating mutant and wild-type TTR levels.45 Phase II placebo-controlled clinical studies in ATTR patients are in progress.
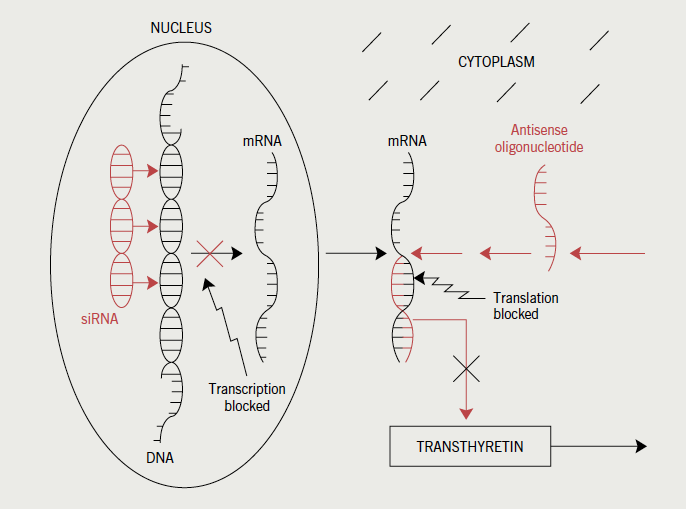
Small-interfering RNA (siRNA) (also known as short-interfering-RNA or silencing-RNA) are double-stranded RNA molecules that cause post-transcriptional gene silencing (figure 4). They are administered in lipid nanoparticles that target wild and mutant TTR synthesis in the liver. In pre-clinical studies, a highly specific siRNA (ALN-TTR) resulted in almost complete regression of human mutant TTR protein accumulation from peripheral tissues of transgenic mice.46 A phase I, multi-national trial conducted in 32 patients with hereditary amyloidosis, has also demonstrated TTR lowering.47
Other proposals as treatments for TTR amyloidosis
A catechin component of green tea (epigallocatechin-3-gallate) has recently been shown, in a small study, to halt the progression of TTR-related cardiomyopathy. After 12 months of consuming green tea, 14 of 19 evaluable patients showed no increase in left ventricular (LV) wall thickness or mass on echo. A smaller subgroup undergoing cardiac magnetic resonance imaging (MRI), all showed a decrease in LV myocardial mass.48 Curcumin, a yellow pigment found in tumeric, has also been demonstrated in mice to considerably increase plasma TTR stability and decrease TTR deposition.49 Another nutraceutical that appears to inhibit TTR amyloidogenesis, by inhibiting transthyretin tetramer dissociation, is the natural product genistein found in soy.50 Genistein is orally active, indicating that patients could benefit from increasing their intake of soy products.
The glycosaminoglycan heparan sulphate appears to be integral to the aggregation of TTR monomer units and may represent a further target for treatments in preventing transthyretin fibrillisation.
As in AL amyloidosis, the compound CPHPC, which binds circulating SAP and eventually removes SAP from amyloid deposits,27 might also be efficacious in ATTR. Subsequent anti-SAP antibody therapy might also be applicable.28
A combination of the anthracine IDOX (4’-iodo-4’-deoxydoxorubicin) and the bile acid TUDCA (tauroursodeoxycholic acid) was recently trialled in 20 patients with ATTR and demonstrated stable neuropathy impairment scores and, importantly, no progression of cardiac involvement.51
Further proposals for therapy include immunisation against specific amyloidogenic TTR variants.52
Senile systemic amyloidosis: wild-type transthyretin amyloid
Senile systemic amyloidosis (SSA) is caused by the deposition of wild-type transthyretin. This predominantly occurs within the heart. SSA is almost exclusively a disease of elderly men, and as such is probably frequently overlooked. While present in around 80% of hearts in those over 80 years,53 it clinically affects around 25% of this age group.54 A recent study of 102 patients with SSA showed that 96% were male, congestive heart failure was the presenting feature in 86% and the median age-adjusted survival (without disease-modifying therapy) was 4.6 years.55 Development of heart failure is slow but insidious and atrial fibrillation is not infrequent. Unlike AL amyloidosis, patients with SSA may tolerate ACE inhibitors or angiotensin receptor blockers. Atrioventricular block will respond to pacing, and strong consideration should be given to bi-ventricular devices. SSA is not associated with other significant organ involvement making heart transplantation a reasonable proposition when there is severe cardiac involvement.56,57
Novel therapies in the treatment of senile systemic amyloidosis
With SSA, the main issue has been identifying these patients, largely compounded by extremes of age and a reluctance by physicians to perform cardiac biopsies. Many of the approaches described for the management of ATTR will also be applicable in SSA. With this in mind, and the potential to use stabilising molecules, including tafamidis and diflunisal, the justification of determining a diagnosis by endomyocardial biopsy has increased. In units familiar with this procedure, the complication rate (perforation, tamponade, valve damage, fistulae formation and arrhythmia) is low, with mortality estimates of 0–0.4%.58 A Japanese report from a total of 214 institutes reported a mortality of 10 cases in 19,964 (0.05%) cardiac biopsy procedures.59
Conclusion
Considerable advances have been made in the treatment of amyloid types affecting the heart. An early diagnosis of the disease, and an exact diagnosis of amyloid type, is critical to achieve any chance of success. Resolution of cardiac amyloid may take several years but patients now have treatment options that may permit this.
Acknowledgement
I am grateful to the organisers and contributors of the International Society of Amyloidosis for the XIIth (2010) and XIIIth (2012) International Symposia on Amyloidosis for the ability to be able to cite their work.
Conflict of interest
SWD has received payment from Johnson & Johnson Pharmaceuticals for studies into the use of Velcade (Bortezomib) and is funded by ISIS Pharmaceuticals in an advisory role on a Data Safety and Monitoring Board for an antisense oligonucleotide study in TTR amyloidosis.
Key messages
- Amyloid deposits can be ‘resorbed’ and organ function (including the heart) can be restored
- Advances in treatment in all three amyloid types support performing cardiac biopsies to determine amyloid type
- Heart transplantation for senile systemic amyloidosis appears a successful approach in patients who present at a sufficiently young age
- Small molecule kinetic stabilisation of transthyretin, preventing conformational change permissive to amyloid assembly, shows particular promise
- The impact of treatment in AL amyloidosis can be measured using serum free light chain estimates
- Serum amyloid P (SAP) scans allow whole body amyloid load to be monitored
- Biomarkers (NT-Pro-BNP and troponin) allow cardiac response to be monitored
References
- Wechalekar AD, Gillmore JD, Gibbs SDJ et al. 25 years of amyloidosis in the UK – single centre experience of 5100 patients. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 28.
- Dubrey SW, Comenzo RL. Amyloid heart disease: current and future therapies. QJM 2012;105:617–31. http://dx.doi.org/10.1093/qjmed/hcr259
- Kristen AV, Dengler TJ, Hegenbart U et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk of sudden cardiac death. Heart Rhythm 2008;5:235–40. http://dx.doi.org/10.1016/j.hrthm.2007.10.016
- Lin G, Dispenzieri A, Grogan M, Kyle R, Brady PA. Outcomes of implantable defibrillators in patients with cardiac amyloidosis. Amyloid 2010;17:166a. http://dx.doi.org/10.3109/13506121003737419
- Falk RH, Dubrey SW. Amyloid heart disease. Progress Cardiovasc Dis 2010;52:347–61. http://dx.doi.org/10.1016/j.pcad.2009.11.007
- Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation 2012;124:1079–85. http://dx.doi.org/10.1161/CIRCULATIONAHA.110.010447
- Fitzgerald B, Bashford J, Scalia G. The return of the normal heart: cardiac amyloidosis disappears after bone marrow transplantation. Eur Heart J 2011;32:184. http://dx.doi.org/10.1016/j.hlc.2013.01.013
- Palladini G, Lavatelli F, Russo P et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood 2006;107:3854–8. http://dx.doi.org/10.1182/blood-2005-11-4385
- Lachmann HJ, Gallimore R, Gillmore JD et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol 2003;122:78–84. http://dx.doi.org/10.1046/j.1365-2141.2003.04433.x
- Skinner M, Sanchorawala V, Seldin DC et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Int Med 2004;140:85–93. http://dx.doi.org/10.7326/0003-4819-140-2-200401200-00008
- Gertz MA, Lacy MQ, Dispenzieri A et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica 2007;92:1415–18. http://dx.doi.org/10.3324/haematol.11413
- Cordes S, Dispenzieri A, Lacy MQ et al. Ten year survival following autologous stem cell transplantation for immunoglobulin light chain amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 38.
- Sanchorawala V, Skinner M, Quillen K et al. Long term outcome of patients with AL amyloidosis treated with high dose melphalan and stem cell transplantation. Blood 2007;110:3561–3. http://dx.doi.org/10.1182/blood-2007-07-099481
- Gertz MA, Lacy MQ, Dispenzieri A et al. Autologous stem cell transplant for immunoglobulin light chain amyloidosis: a status report. Leuk Lymphoma 2010;51:181–7. http://dx.doi.org/10.3109/10428194.2010.524329
- Meier-Ewert HK, Sanchorawala V, Berk J et al. Regression of cardiac wall thickness following chemotherapy and stem-cell transplantation for AL amyloidosis. Amyloid 2010;17:150a. http://dx.doi.org/10.3109/13506129.2011.574354048
- Madan S, Kumar S, Dispenzieri A et al. Outcomes with high-dose therapy (HDT) and peripheral blood (PB) stem cell transplantation for AL amyloidosis with cardiac involvement. Amyloid 2010;17:182a. http://dx.doi.org/10.3109/13506121003737419
- Jaccard A, Moreau P, Leblond V et al. High dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med 2007;357:1083–93. http://dx.doi.org/10.1056/NEJMoa070484
- Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011;4:47–53. http://dx.doi.org/10.1186/1756-8722-4-47
- Kastritis E, Wechalekar AD, Dimopoulos MA et al. Bortezomib with or without dexamethasone in primary (light chain) systemic amyloidosis. J Clin Oncol 2010;28:1031–7. http://dx.doi.org/10.1200/JCO.2009.23.8220
- Dubrey SW, Reece DE, Sanchorawala V et al. Bortezomib in a phase 1 trial for patients with relapsed AL amyloidosis: cardiac responses and overall effects. QJM 2011;104:957–70. http://dx.doi.org/10.1093/qjmed/hcr105
- Venner CP, Lane T, Foard D et al. Cyclophosphamide, bortezomib and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood 2012;119:4387–90. http://dx.doi.org/10.1182/blood-2011-10-388462
- Jaccard A, Comenzo RL, Wechalekar AD et al. Efficacy of bortezomib/cyclophosphamide, dexamethasone (VCD) chemotherapy in naïve patients with high risk cardiac light chain amyloidosis (Mayo clinic stage III). XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP62.
- Landau H, Hassoun H, Rosenzweig MA et al. Improved hematologic responses following risk adapted stem cell transplant (SCT) and bortezomib consolidation in systemic light-chain amyloidosis (AL) is associated with long term organ improvement. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 45.
- Wechalekar AD, Goodman HJB, Lachmann HJ, Offer M, Hawkins PN, Gillmore JD. Safety and efficacy of risk-adapted cyclophosphamide, thalidomide, and dexamethasone in systemic AL amyloidosis. Blood 2007;109:457–64. http://dx.doi.org/10.1182/blood-2006-07-035352
- Kumar SK, Hayman SR, Buadi FK et al. Lenalidomide, cyclophosphamide and dexamethasone (cRd) for light chain amyloidosis: long term results from a Phase 2 trial. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 44.
- Dispenzieri A, Buadi F, Lauermann K et al. The activity of pomalidomide in patients with immunoglobulin light chain amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP 64.
- Pepys MB, Herbert J, Hutchinson WL et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 2002;417:254–9. http://dx.doi.org/10.1038/417254a
- Bodin K, Ellmerich S, Kahan MC et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature 2012;468:93–7. http://dx.doi.org/10.1038/nature09494
- Sattianayagam PT, Gibbs SDJ, Pinney JH et al. Solid organ transplantation in AL amyloidosis. Am J Transplant 2010;10:2124–31. http://dx.doi.org/10.1111/j.1600-6143.2010.03227.x
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis and referral. Heart 2011;97:75–84. http://dx.doi.org/10.1136/hrt.2009.190405
- Jacobson DR, Pan T, Kyle RA, Buxbaum JN. Transthyretin ILE20, a new variant associated with late-onset cardiac amyloidosis. Hum Mutat 1997;9:83–5. http://dx.doi.org/10.1002/(SICI)1098-1004(1997)9:1<83::AID-HUMU19>3.0.CO;2-L
- Dungu J, O’Donnell M, Hawkins PN, Anderson LJ. Systematic review of 1142 admissions with acute heart failure reveals high frequency of transthyretin V122I cardiac amyloidosis in Afro-Caribbeans patients. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP49.
- Connors LH, Prokaeva T, Lim A et al. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J 2009;158:607–14. http://dx.doi.org/10.1016/j.ahj.2009.08.006
- Noumi B, Latif F, Helmke S et al. Differences between transthyretin (ATTR) cardiac amyloidosis secondary to V122I mutations and wild type TTR presenting to a tertiary referral center. J Card Fail 2010;16:S40. http://dx.doi.org/10.1016/j.cardfail.2010.06.138
- Wilczek HE, Larsson M, Ericzon BG. Long term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid 2011;Suppl 1:193–5. http://dx.doi.org/10.3109/13506129.2011.574354072
- Yazaki M, Mitsuhashi S, Tokuda T et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant 2007;7:235–42. http://dx.doi.org/10.1111/j.1600-6143.2006.01585.x
- Suhr OB, Ericzon BG, Friman S. Long term follow up of survival of liver transplant recipients with familial amyloid polyneuropathy (Portuguese type). Liver Transpl 2002;8:787–94. http://dx.doi.org/10.1053/jlts.2002.34386
- Alhamadsheh MM, Connelly S, Cho A et al. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med 2011;3:97ra81. http://dx.doi.org/10.1126/scitranslmed.3002473
- Coelho T, Maia L, Martins da Silva A et al. Tafamidis (Fx-1006A): a first-in-class disease-modifying therapy for transthyretin familial amyloid. Amyloid 2010;17(suppl 1):OP-066.
- Coelho T, Maia L, Martins da Solva A et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized controlled trial. Neurology 2012;79:785–92. http://dx.doi.org/10.1212/WNL.0b013e3182661eb1
- Merlini G, Coelho T, Falk RH et al. Transthyretin stabilisation, efficacy and safety of tafamidis for the treatment of transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 49.
- Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 2006;13:236–49. http://dx.doi.org/10.1080/13506120600960882
- Berk JL, Obici L, Zeldenrust SR et al. The diflunisal trial: demographics, baseline neurologic staging, and adverse events. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP69.
- Whelan CJ, Sattianayagam P, Dungu J et al. Tolerability of diflunisal therapy in patients with transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP56.
- Ackermann EJ, Guo S, Booten S et al. Clinical development of an anti-sense therapy for the treatment of hereditary transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP73.
- Sah DWY, Chen Q, Costelha S et al. Pre-clinical development of ALN-TTR, a novel RNAi therapeutic for the treatment of transthyretin amyloidosis. Available from: http:///www.alnylam.com/Files/Presentations/ALNY-PNS-TTR-June2011.pdf
- Coelho T, Silva A, Suhr OB et al. Final phase 1 safety, pharmacokinetic and pharmacodynamic results for ALN-TTR01, a novel RNAi therapeutic for the treatment of transthyretin amyloidosis. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP 74.
- Kristen AV, Buss S, Mereles D et al. Green tea halts progression of cardiac transthyretin amyloidosis – a pilot study. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. PC 43.
- Ferreira N, Saraiva MJ, Almeida MR. Curcumin as a novel natural compound acting as TTR amyloidosis inhibitor in vivo. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP 70.
- Green NS, Foss TR, Kelly JW. Genistein, a natural product from soy, is a potent inhibitor of transthyretin amyloidosis. Proc Nat Acad Sci USA 2005;102:14545–50. http://dx.doi.org/10.1073/pnas.0501609102
- Merlini G, Obici L, Cortese A, Saraiva MJ. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. In: Ando Y (ed.). VIIIth International Symposium on Familial Amyloid Polyneuropathy. Kumamoto, Japan: Intractable Diseases Foundation, 2011;58.
- Nuvolone M, Obici L, Merlini G. Transthyretin-associated familial amyloid polyneuropathy – current and emerging therapies. US Neurology 2012;8:24–32. Available from: http://www.touchneurology.com/articles/transthyretin-associated-familial-amyloid-polyneuropathy-current-and-emerging-therapies-0
- Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med 2005;165:1425–9. http://dx.doi.org/10.1001/archinte.165.12.1425
- Tanskanen M, Peuralinna T, Polvikoski T et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha-2 macroglobulin and tau: a population based study. Ann Med 2008;40:232–9. http://dx.doi.org/10.1080/07853890701842988
- Connors LH, Sam F, Doros G et al. Senile systemic amyloidosis: a large cohort study detailing clinical features, laboratory results and survival. XIIIth International Symposium on Amyloidosis, 6–10 May 2012, Groningen, Netherlands. OP 48.
- Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant 2004;23:1142–53. http://dx.doi.org/10.1016/j.healun.2003.08.027
- Roig E, Almenar L, Gonzalez-Vilchez F et al. Outcomes of heart transplantation for cardiac amyloidosis: subanalysis of the Spanish registry for heart transplantation. Am J Transplant 2009;9:1414–19. http://dx.doi.org/10.1111/j.1600-6143.2009.02643.x
- Kilo J, Laufer G, Antretter H. Endomyocardial biopsy – jugular/subclavian vein approach. Multimed Man Cardiothorac Surg 2006;published online. http://dx.doi.org/10.1510/mmcts.2005.001149
- Hiramitsu S, Hiroe M, Morimoto S-I et al. Safety of cardiac biopsy based on a 32 year experience in Japan. J Am Coll Cardiol 1995;25(suppl 1):166A. http://dx.doi.org/10.1016/0735-1097(95)92143-S
