
Case presentation
An 83-year-old man presented with progressive shortness of breath and moderate lower extremity oedema over a period of several months. His past medical history is notable for hypertension, non-insulin dependent diabetes, bilateral carpal tunnel syndrome and aortic stenosis. At the time of presentation he was found to have new-onset atrial flutter with rapid ventricular response. An echocardiogram demonstrated moderate concentric left ventricular hypertrophy and moderate aortic stenosis with an ejection fraction of 55% and mildly reduced right ventricular function. A left heart catheterisation confirmed the presence of moderate aortic stenosis. He underwent successful cardioversion with restoration of sinus rhythm, however, his symptoms only mildly improved. He continued to experience progressive heart failure symptoms with minimal exertion and recurrent atrial flutter, as well as recurrent pleural effusions, requiring high-dose diuretics, recurrent thoracenteses, and atrial flutter ablation. At this time his symptoms were thought to be disproportionate to the degree of aortic stenosis and his atrial arrhythmias, and he was referred for evaluation of cardiac amyloidosis.
Clinical suspicion and ‘red-flag’ findings
Although recognition of ATTR-CA as a cause of heart failure has increased, reaching an accurate diagnosis remains elusive and the physician must maintain a high index of clinical suspicion in whom to consider further evaluation for ATTR-CA. Patients most frequently present with multi-system involvement to providers from various medical subspecialties, rather than isolated cardiac involvement, necessitating an algorithmic approach to anyone with suspected cardiac amyloidosis.3 First and foremost, patients should exhibit concentric left ventricular hypertrophy with resultant diastolic dysfunction, frequently along with a history of heart failure. Several important features, commonly termed ‘red flags’, represent important clues for ATTR-CA, which are highlighted in table 1.4
Table 1. Clinical and imaging features prompting heightened suspicion for cardiac amyloidosis
| The presence of concentric left ventricular hypertrophy in the absence of a history of hypertension (or disproportionate to the degree of hypertension) |
| Bilateral carpal tunnel syndrome and/or lumbar stenosis |
| Elderly patients presenting with a new ‘diagnosis’ of hypertrophic cardiomyopathy |
| Intolerance to beta blockers, calcium channel blockers, ACE inhibitors, and angiotensin receptor blockers |
| Polyneuropathy and/or autonomic insufficiency |
| Low flow, flow gradient severe aortic stenosis |
| Discordant echocardiographic mass and QRS voltage on electrocardiography |
| Reduced global longitudinal with apical ‘sparing’ |
| Diffuse late gadolinium enhancement on cardiac MRI |
| Elevated circulating plasma levels of natriuretic peptides |
| Persistently elevated cardiac troponin levels |
| Key: ACE = angiotensin-converting enzyme; MRI = magnetic resonance imaging |
Clinical features
Importantly, the left ventricular hypertrophy is either unexplained or often is out-of-proportion to the degree of hypertension. In fact, patients with a history of hypertension often exhibit ‘improvement’ in their hypertension with resultant down-titration of their antihypertensive regimen. Orthostatic hypotension along with intolerance to beta blockers, calcium channel blockers, angiotensin-converting enzyme (ACE) inhibitors and angiotensin-receptor blockers (ARBs) commonly occurs. In addition, atrial arrhythmias and/or atrioventricular block in the presence of heart failure with preserved ejection fraction (HFpEF) are common findings, particularly in elderly patients with wtATTR.5-9 Furthermore, particular attention should be paid to elderly patients with severe aortic stenosis, particularly low-flow, low-gradient aortic stenosis, as 16–33% of subjects undergoing transcatheter aortic valve replacement have concomitant ATTR-CA.10,11
Owing to the systemic nature of amyloid deposition, CA can importantly manifest as either carpal tunnel syndrome or lumbar stenosis with the presence of bilateral carpal tunnel syndrome being a potential ‘warning sign’, which can present up to 10 years prior to the presence of CA.12,13 Sperry et al. demonstrated that 10.2% of patients undergoing carpal tunnel release had histopathologic evidence of amyloid.13 Additionally, given the predilection for nervous and autonomic system involvement, concurrent HFpEF with polyneuropathy and/or dysautonomia should further raise suspicion.14
Echocardiography
The echocardiogram remains the cornerstone ‘screening’ test, often resulting in a heightened suspicion for an underlying infiltrative cardiomyopathy. It is important to remember that individual echocardiographic features of CA are nonspecific in nature and disorders such as hypertensive heart disease, hypertrophic cardiomyopathy, and Fabry’s disease can present with similar echocardiographic findings but, in totality, can be suggestive of CA.15 In addition, there is no reliable means of differentiation of AL-CA from ATTR-CA based on echocardiography alone.
Classically, patients with CA often demonstrate concentric left ventricular hypertrophy with septal and posterior wall thickness of ≥12 mm (figures 1A and 1B).16 This, along with other two-dimensional characteristics including atrial enlargement, hypertrophy of the right ventricular free-wall, thickened atrioventricular valves and the presence of a pericardial effusion should heighten suspicion.17 Additionally, the myocardium may present with a ‘speckled’ appearance due to the echogenic nature of amyloid within the myocardium.18,19 Particular caution should be paid to this finding; however, as myocardial speckling can be readily seen in the harmonic imaging mode in normal myocardial tissue.16,20 Fundamental imaging should be used to evaluate for the presence of myocardial speckling.
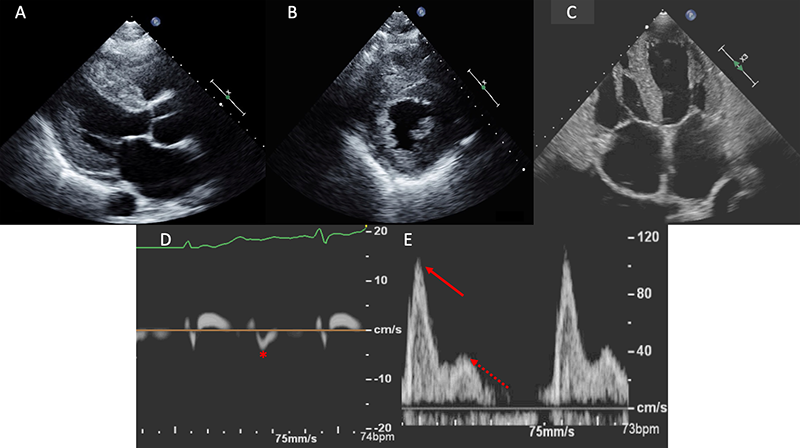
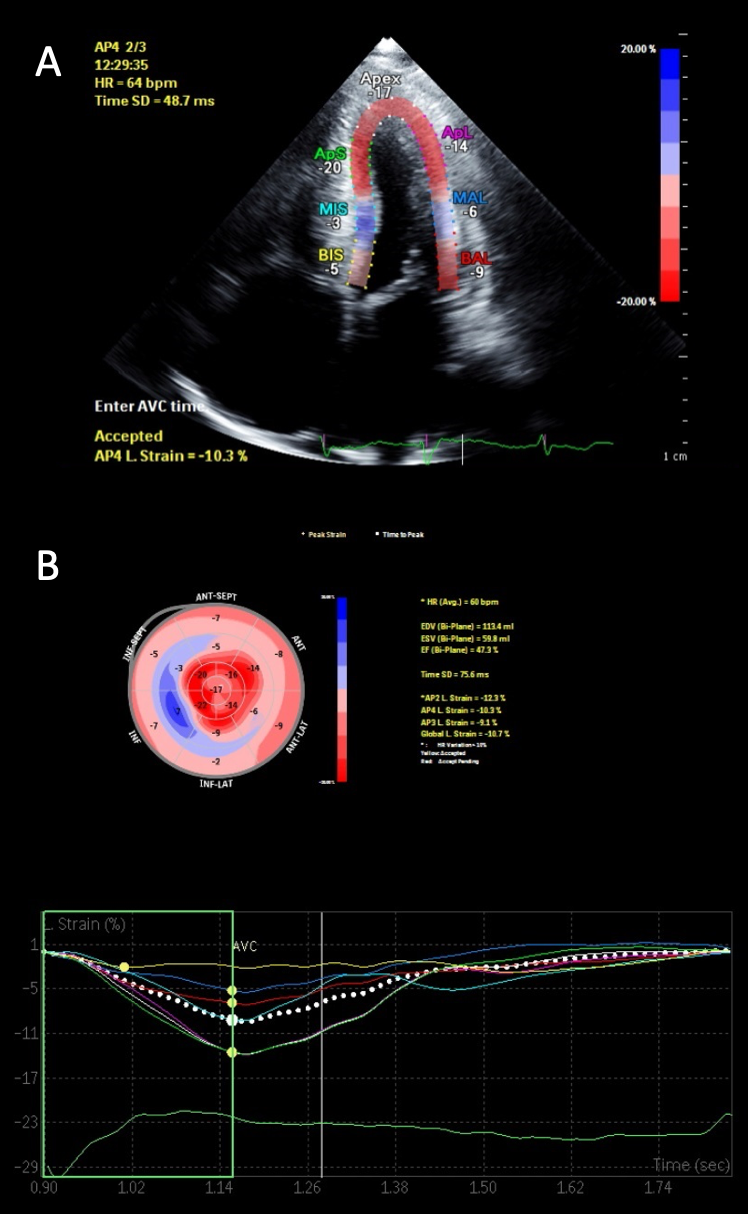
Diastolic function is nearly universally adversely impacted with the ultimate progression to restrictive physiology in advanced stages.21 This is demonstrated in figure 1, with the typical echocardiographic features of restrictive diastolic filling. Although CA has classically been described as presenting with a preserved ejection fraction, this, too, is dependent upon the disease stage. Global longitudinal strain (GLS) is a powerful tool as the pattern typically demonstrates an ‘apical-sparing’ pattern with a base-to-apex strain gradient, and GLS will decline prior to any decline in left ventricular ejection fraction.22 As seen in figure 2, this ‘apical-sparing’ pattern can be represented using a ‘bull’s-eye’ plot of the heart. This can help distinguish CA from other mimickers that may result in left ventricular hypertrophy.23
Cardiac magnetic resonance
Cardiac magnetic resonance (CMR) provides both structural information and tissue characterisation due to superb spatial resolution with a high signal-to-noise ratio resulting in excellent image quality. Importantly, similar to echocardiography, CMR does not distinguish between AL-CA and ATTR-CA and additional diagnostic evaluation is mandatory to obtain an accurate diagnosis. Classically, amyloidosis is described as having a pattern of global subendocardial late gadolinium enhancement (LGE) on CMR, however, this can also be transmural, diffuse or patchy leading to challenges in reaching the accurate diagnosis.24,25 Importantly, false-negative results can be obtained if myocardial tissue is not correctly ‘nulled’ and careful attention needs to be paid when choosing an appropriate inversion time so that the myocardium can be accurately nulled for scan interpretation.26
Electrocardiography
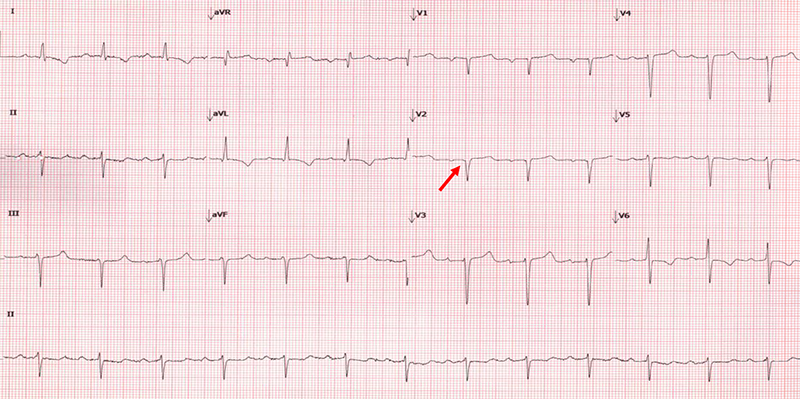
Traditionally, it was thought that a low-voltage electrocardiogram (ECG) was compelling evidence for the diagnosis of CA. In practice, however, this finding is an insensitive measure to screen for CA. A more sensitive ECG sign for the presence of CA is the discordance between echocardiographic mass and QRS voltage, which can be represented by a voltage/mass formula, with reported sensitivities as high as 80%.27,28 Frequently, patients with CA will be incorrectly diagnosed with coronary disease due to pseudo-infarction patterns (figure 3) in the precordial leads, and this ECG finding has been described in nearly 70% of subjects with CA.28
Biomarkers
Serum troponin levels tend to be mildly elevated in patients with CA, postulated to be secondary to direct cellular toxicity and microvascular ischaemia, while circulating plasma levels of natriuretic peptides (BNP and NT-proBNP) are often disproportionately elevated when compared with their clinical presentation, frequently disproportionate to the degree of clinical heart failure. Suggested mechanisms include increased intracardiac pressure and direct cellular or mechanical effect of amyloid fibrils on natriuretic peptide expression in cardiac cells.4,29-31
Diagnosis of ATTR-CA
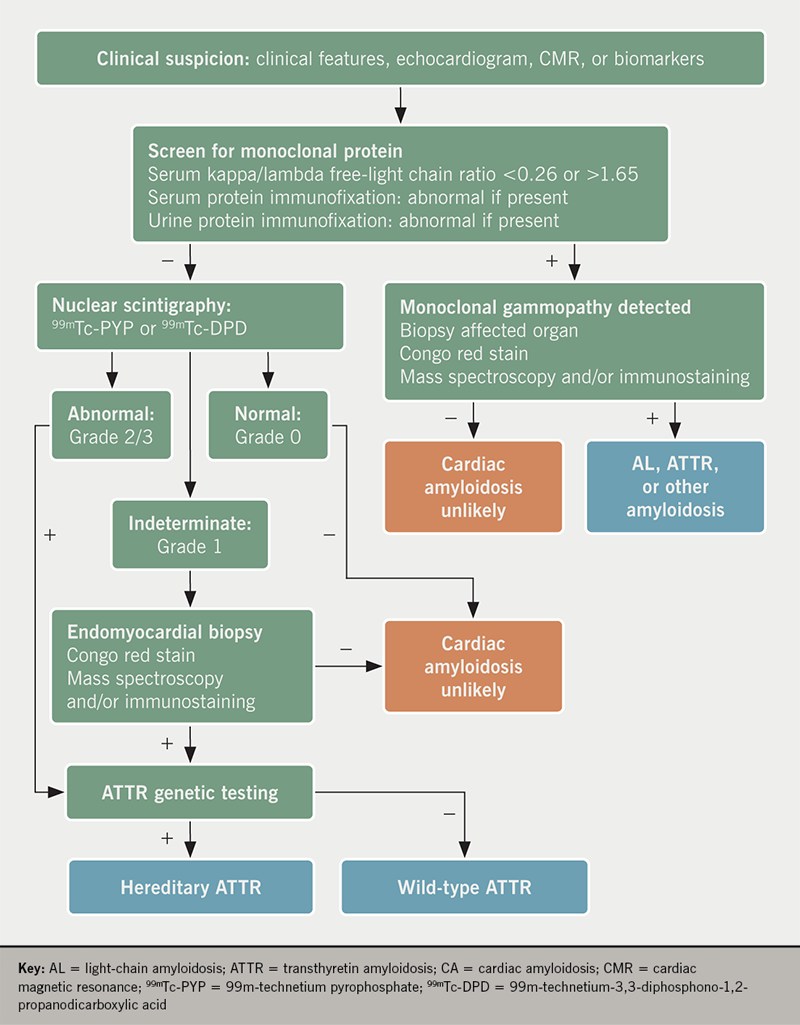
Once suspected on the basis of prior clinical characteristics, imaging findings, and/or cardiac biomarkers, it is paramount to either confirm or refute the diagnosis of ATTR-CA, particularly as disease-modifying therapies emerge in the treatment of ATTR-CA.32 More specifically, prompt recognition and early diagnosis of ATTR-CA is critical in decreasing morbidity and mortality in this patient population. We present a diagnostic framework in the evaluation of patients with suspected cardiac amyloidosis using both nuclear scintigraphy alongside serum and urine analyses for AL-CA, highlighted in figure 4.
Evaluation for monoclonal gammopathy
Every patient with suspected CA must be excluded for the presence of AL-CA, as delays in diagnosis can have significant prognostic implications. Traditionally, serum protein electrophoresis has been used to evaluate for plasma cell dyscrasias, however, this method alone is an insensitive measure.3 Rather, the combination of serum and urine immunofixation and quantification of serum free light chains have 99% sensitivity for the diagnosis of AL-CA. A serum kappa/lambda free-light chain ratio of either <0.26 or >1.65, or the presence of an M-spike on immunofixation, is considered abnormal.33,34
Haematology consultation is encouraged in the presence of an M protein or an abnormal light-chain ratio. Note should be made that monoclonal gammopathy of undetermined significance (MGUS) may be incorrectly diagnosed as AL-CA, particularly in the elderly population. Similarly, up to 50% of subjects with ATTR-CA have concurrent MGUS, making it paramount to correctly identify the underlying disease process.35 Furthermore, renal insufficiency can lead to an elevated kappa to lambda ratio and expert consultation is encouraged.33,34
Nuclear scintigraphy
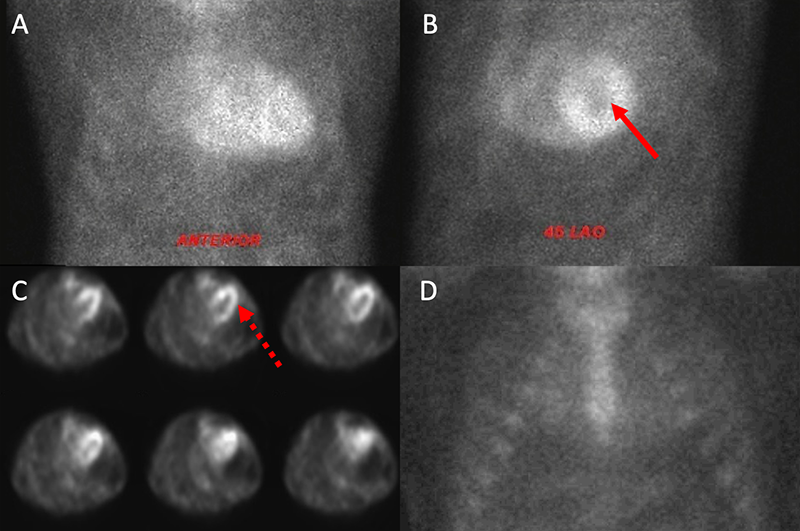
Myocardial scintigraphy using 99mTc-phosphate derivatives has emerged as a highly sensitive and specific non-invasive imaging technique for the accurate diagnosis of ATTR-CA.36-38 Importantly, these bone tracers are particularly avid for ATTR-CA (both wtATTR and hATTR) while they demonstrate minimal to no uptake in AL-CA. Specifically, 99m-technetium pyrophosphate (99mTc-PYP) is used for detection of ATTR-CA in the United States and 99m-technetium-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) has shown promise in Europe.39,40 The exact mechanism for uptake is unknown, however, it is presumed to be calcium-mediated.15,38 When a circular region of interest (ROI) is made around the heart (to represent myocardial 99mTc-PYP uptake) and this is compared with a ‘mirror image’ over the contralateral chest, a heart/contralateral lung (H/CL) ratio of >1.5 has been shown to be 97% sensitive and 100% specific in the identification of ATTR-CA.36 A semi-quantitative visual scoring system of myocardial uptake developed by Perugini et al. is an additional means of identification of ATTR-CA with an excellent sensitivity (>99%) for detection of ATTR-CA.39 This intense myocardial uptake is demonstrated in figures 5A and 5B. Myocardial uptake can be further confirmed using myocardial single-photon emission computed tomography (SPECT) (figure 5C). Importantly, AL-CA can occasionally result in a mild grade 1 uptake (or potentially higher), and; therefore, laboratory testing to exclude a monoclonal protein must be pursued when obtaining myocardial scintigraphy to avoid false-positive results.41
Tissue biopsy
Despite recent advances in nuclear scintigraphy, endomyocardial biopsy remains the gold standard for diagnosis of CA with high sensitivity and specificity.42 Apple-green birefringence is visualised under polarised light with Congo red staining. Proteomic and mass spectrometry technology is used to determine particular amyloid proteins.43 While biopsy is discouraged as a first-line diagnostic strategy, it remains useful in clarifying the diagnosis when nuclear scintigraphy is indeterminate and laboratory testing has excluded AL-CA. Furthermore, in cases of abnormal laboratory testing for AL-CA along with grade 1 or greater uptake on nuclear scintigraphy, biopsy is necessary to further subtype. Despite its role in diagnostic clarification, cardiac biopsy is not widely available at many centres, and remains an invasive procedure with rare, yet serious, potential complications.44 The diagnostic yield of extra-cardiac tissue depends on the underlying aetiology of the amyloidosis. Generally, a fat-pad biopsy is not a recommended strategy for the diagnosis of ATTR-CA due to its low sensitivity.45 The overall sensitivity for AL amyloidosis is approximately 79–84%, while it is much lower in ATTR amyloidosis (hATTR: 45–67%; wtATTR: 14–15%).45-47
Genotyping
Once a diagnosis of ATTR-CA is established, TTR gene sequencing is recommended in all patients as this is the only method of differentiation of hATTR from wtATTR. While hATTR is inherited in an autosomal dominant fashion, there is significant variability in the inheritance pattern. Depending on the mutation, there can be varying degrees of neuropathies, cardiac involvement and ages of presentation. With the multitude of various mutations, a clinical spectrum exists, ranging from predominantly neurologic symptoms with early onset to predominantly cardiac symptoms with later onset.48 For instance, an endemic Val30Met mutation found in patients of Portuguese and Japanese descent presents at a younger age with a predominant sensorimotor polyneuropathy.49 On the other hand, the Val122Ile mutation is prevalent in the African-American population and tends to present with predominant cardiac involvement.50
Disease staging
The combination of circulating natriuretic peptide levels and troponin levels can be prognostically significant. The Mayo Clinic developed a staging system for wtATTR on the basis of NT-proBNP and troponin reaching thresholds of 3,000 pg/ml and 0.05 ng/ml, respectively. These are as follows:
- Stage I when both biomarkers are below these diagnostic criteria (median survival 66 months)
- Stage II when either is above this diagnostic threshold (median survival 42 months)
- Stage III when both are elevated (median survival 20 months).8
The UK National Amyloidosis Centre ATTR staging system categorises patients according to the same NT-proBNP threshold of 3,000 pg/ml and/or having an estimated glomerular filtration rate <45 ml/min with stages assigned if patients had reached neither threshold, either, or both. Accordingly, Stage I patients had a mean survival of 69 months while the mean survival of Stage II and III patients were 47 months and 24 months, respectively.51 Lastly, while no specific echocardiographic parameters are predictors of survival, CMR has demonstrated promise in assessment of amyloid burden through T1 mapping and myocardial extracellular volume fraction.52,53
Clinical approach to the patient
A repeat echocardiogram was performed and there were several features consistent with CA. There was moderate concentric left ventricular hypertrophy with a myocardial thickness of 14 mm along with a mildly reduced ejection fraction, bi-atrial enlargement, a restrictive filling pattern with a high E/A ratio and significantly reduced early mitral tissue Doppler velocity of 0.04 cm/s (figures 1C–E). Although there was evidence of moderate aortic stenosis with a calculated aortic valve area of 1.2 cm2 and a mean gradient of 29 mmHg, his symptoms were disproportionate to the degree of aortic stenosis. Given the echocardiographic findings, the presence of bilateral carpal tunnel syndrome and the patient’s older age, the presentation appeared to be consistent with ATTR-CA. As a first step, AL-CA was excluded via serum and urine immunofixation along with quantification of serum free light chains. Nuclear scintigraphy subsequently was performed, confirming a diagnosis of ATTR-CA with intensive myocardial uptake on both planar and SPECT imaging at one hour (figures 3A-C). Transthyretin gene sequencing did not show evidence of any mutant proteins, thus yielding a diagnosis of wtATTR.
Key messages
- Transthyretin cardiac amyloidosis is a common and widely underdiagnosed cause of heart failure, particularly in the elderly
- There are various clinical and echocardiographic clues that should raise the suspicion for the presence of cardiac amyloidosis
- Nuclear scintigraphy with either 99mTc-PYP or 99mTc-DPD is diagnostic for the presence of transthyretin cardiac amyloidosis
- All patients undergoing evaluation for transthyretin cardiac amyloidosis need to undergo concurrent laboratory testing to exclude the presence of light-chain amyloidosis
Conflicts of interest
JMK and GP have received personal fees from Pfizer Inc. RK, RV, AR: none declared.
References
1. Maurer MS, Hanna M, Grogan M et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol 2016;68:161–72. https://doi.org/10.1016/j.jacc.2016.03.596
2. Pinney JH, Whelan CJ, Petrie A et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc 2013;2:e000098. https://doi.org/10.1161/JAHA.113.000098
3. Rapezzi C, Lorenzini M, Longhi S et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev 2015;20:117–24. https://doi.org/10.1007/s10741-015-9480-0
4. Witteles RM, Bokhari S, Damy T et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail 2019;7:709–16. https://doi.org/10.1016/j.jchf.2019.04.010
5. Rapezzi C, Merlini G, Quarta CC et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 2009;120:1203–12. https://doi.org/10.1161/CIRCULATIONAHA.108.843334
6. Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Heart Fail 2018;5:772–9. https://doi.org/10.1002/ehf2.12308
7. Connors LH, Sam F, Skinner M et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation 2016;133:282–90. https://doi.org/10.1161/CIRCULATIONAHA.115.018852
8. Grogan M, Scott CG, Kyle RA et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol 2016;68:1014–20. https://doi.org/10.1016/j.jacc.2016.06.033
9. Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG. Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch Intern Med 1995;155:469–73. https://doi.org/10.1001/archinte.1995.00430050045005
10. Castaño A, Narotsky DL, Hamid N et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J 2017;38:2879–87. https://doi.org/10.1093/eurheartj/ehx350
11. Calero Núñez S, Tercero Martínez A, García López JC, Jiménez-Mazuecos J. [Wild-type transthyretin-related cardiac amyloidosis and degenerative aortic stenosis: two inter-related pathologies in the elderly]. Rev Esp Geriatr Gerontol 2017;52:167–70. https://doi.org/10.1016/j.regg.2016.05.002
12. Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin-derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci 2014;119:223–8. https://doi.org/10.3109/03009734.2014.895786
13. Sperry BW, Reyes BA, Ikram A et al. Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J Am Coll Cardiol 2018;72:2040–50. https://doi.org/10.1016/j.jacc.2018.07.092
14. Gertz MA, Benson MD, Dyck PJ et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol 2015;66:2451–66. https://doi.org/10.1016/j.jacc.2015.09.075
15. Maurer MS, Elliott P, Comenzo R, Semigran M, Rapezzi C. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation 2017;135:1357–77. https://doi.org/10.1161/CIRCULATIONAHA.116.024438
16. Mohty D, Damy T, Cosnay P et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis 2013;106:528–40. https://doi.org/10.1016/j.acvd.2013.06.051
17. Damy T, Maurer MS, Rapezzi C et al. Clinical, ECG and echocardiographic clues to the diagnosis of TTR-related cardiomyopathy. Open Heart 2016;3:e000289. https://doi.org/10.1136/openhrt-2015-000289
18. Bhandari AK, Nanda NC. Myocardial texture characterization by two-dimensional echocardiography. Am J Cardiol 1983;51:817–25. https://doi.org/10.1016/S0002-9149(83)80139-8
19. Rahman JE, Helou EF, Gelzer-Bell R et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004;43:410–15. https://doi.org/10.1016/j.jacc.2003.08.043
20. Agha AM, Parwani P, Guha A et al. Role of cardiovascular imaging for the diagnosis and prognosis of cardiac amyloidosis. Open Heart 2018;5:e000881. https://doi.org/10.1136/openhrt-2018-000881
21. Knight DS, Zumbo G, Barcella W et al. Cardiac structural and functional consequences of amyloid deposition by cardiac magnetic resonance and echocardiography and their prognostic roles. JACC Cardiovasc Imaging 2019;12:823–33. https://doi.org/10.1016/j.jcmg.2018.02.016
22. Buss SJ, Emami M, Mereles D et al. Longitudinal left ventricular function for prediction of survival in systemic light-chain amyloidosis: incremental value compared with clinical and biochemical markers. J Am Coll Cardiol 2012;60:1067–76. https://doi.org/10.1016/j.jacc.2012.04.043
23. Phelan D, Collier P, Thavendiranathan P et al. Relative apical sparing of longitudinal strain using two-dimensional speckle-tracking echocardiography is both sensitive and specific for the diagnosis of cardiac amyloidosis. Heart 2012;98:1442–8. https://doi.org/10.1136/heartjnl-2012-302353
24. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol 2007;50:2101–10. https://doi.org/10.1016/j.jacc.2007.08.028
25. Syed IS, Glockner JF, Feng D et al. Role of cardiac magnetic resonance imaging in the detection of cardiac amyloidosis. JACC Cardiovasc Imaging 2010;3:155–64. https://doi.org/10.1016/j.jcmg.2009.09.023
26. Fontana M, Pica S, Reant P et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015;132:1570–9. https://doi.org/10.1161/CIRCULATIONAHA.115.016567
27. Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol 1982;49:9–13. https://doi.org/10.1016/0002-9149(82)90270-3
28. Cyrille NB, Goldsmith J, Alvarez J, Maurer MS. Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am J Cardiol 2014;114:1089–93. https://doi.org/10.1016/j.amjcard.2014.07.026
29. Nordlinger M, Magnani B, Skinner M, Falk RH. Is elevated plasma B-natriuretic peptide in amyloidosis simply a function of the presence of heart failure? Am J Cardiol 2005;96:982–4. https://doi.org/10.1016/j.amjcard.2005.05.057
30. Palladini G, Campana C, Klersy C et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003;107:2440–5. https://doi.org/10.1161/01.CIR.0000068314.02595.B2
31. Perfetto F, Bergesio F, Emdin M, Cappelli F. Troponins in cardiac amyloidosis: multipurpose markers. Nat Rev Cardiol 2014;11:179. https://doi.org/10.1038/nrcardio.2013.129-c1
32. Maurer MS, Schwartz JH, Gundapaneni B et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16. https://doi.org/10.1056/NEJMoa1805689
33. Dispenzieri A, Katzmann JA, Kyle RA et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010;375:1721–8. https://doi.org/10.1016/S0140-6736(10)60482-5
34. Gertz MA. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am J Hematol 2016;91:947–56. https://doi.org/10.1002/ajh.24433
35. Phull P, Sanchorawala V, Connors LH et al. Monoclonal gammopathy of undetermined significance in systemic transthyretin amyloidosis (ATTR). Amyloid 2018;25:62–7. https://doi.org/10.1080/13506129.2018.1436048
36. Bokhari S, Castaño A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging 2013;6:195–201. https://doi.org/10.1161/CIRCIMAGING.112.000132
37. Gertz MA, Brown ML, Hauser MF, Kyle RA. Utility of technetium Tc 99m pyrophosphate bone scanning in cardiac amyloidosis. Arch Intern Med 1987;147:1039–44. https://doi.org/10.1001/archinte.1987.00370060035007
38. Falk RH, Quarta CC, Dorbala S. How to image cardiac amyloidosis. Circ Cardiovasc Imaging 2014;7:552–62. https://doi.org/10.1161/CIRCIMAGING.113.001396
39. Perugini E, Guidalotti PL, Salvi F et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol 2005;46:1076–84. https://doi.org/10.1016/j.jacc.2005.05.073
40. Rapezzi C, Quarta CC, Guidalotti PL et al. Usefulness and limitations of 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy in the aetiological diagnosis of amyloidotic cardiomyopathy. Eur J Nucl Med Mol Imaging 2011;38:470–8. https://doi.org/10.1007/s00259-010-1642-7
41. Gillmore JD, Maurer MS, Falk RH et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016;133:2404–12. https://doi.org/10.1161/CIRCULATIONAHA.116.021612
42. Ardehali H, Qasim A, Cappola T et al. Endomyocardial biopsy plays a role in diagnosing patients with unexplained cardiomyopathy. Am Heart J 2004;147:919–23. https://doi.org/10.1016/j.ahj.2003.09.020
43. Dungu JN. Cardiac amyloid – an update. Eur Cardiol 2015;10:113–17. https://doi.org/10.15420/ecr.2015.10.2.113
44. Deckers JW, Hare JM, Baughman KL. Complications of transvenous right ventricular endomyocardial biopsy in adult patients with cardiomyopathy: a seven-year survey of 546 consecutive diagnostic procedures in a tertiary referral center. J Am Coll Cardiol 1992;19:43–7. https://doi.org/10.1016/0735-1097(92)90049-S
45. Garcia Y, Collins AB, Stone JR. Abdominal fat pad excisional biopsy for the diagnosis and typing of systemic amyloidosis. Hum Pathol 2018;72:71–9. https://doi.org/10.1016/j.humpath.2017.11.001
46. Quarta CC, Gonzalez-Lopez E, Gilbertson JA et al. Diagnostic sensitivity of abdominal fat aspiration in cardiac amyloidosis. Eur Heart J 2017;38:1905–08. https://doi.org/10.1093/eurheartj/ehx047
47. Fine NM, Arruda-Olson AM, Dispenzieri A et al. Yield of noncardiac biopsy for the diagnosis of transthyretin cardiac amyloidosis. Am J Cardiol 2014;113:1723–7. https://doi.org/10.1016/j.amjcard.2014.02.030
48. Rapezzi C, Quarta CC, Obici L et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 2013;34:520–8. https://doi.org/10.1093/eurheartj/ehs123
49. Coelho T, Maurer MS, Suhr OB. THAOS – The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin 2013;29:63–76. https://doi.org/10.1185/03007995.2012.754348
50. Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012;126:1286–300. https://doi.org/10.1161/CIRCULATIONAHA.111.078915
51. Gillmore JD, Damy T, Fontana M et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018;39:2799–806. https://doi.org/10.1093/eurheartj/ehx589
52. Karamitsos TD, Piechnik SK, Banypersad SM et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 2013;6:488–97. https://doi.org/10.1016/j.jcmg.2012.11.013
53. Banypersad SM, Sado DM, Flett AS et al. Quantification of myocardial extracellular volume fraction in systemic AL amyloidosis: an equilibrium contrast cardiovascular magnetic resonance study. Circ Cardiovasc Imaging 2013;6:34–9. https://doi.org/10.1161/CIRCIMAGING.112.978627
