Introduction
A roundtable meeting of experts in pulmonary hypertension was held at the Royal Society, London, in December 2014. The aim was to provide an update on current approaches to the diagnosis and treatment of pulmonary hypertension (PH) in the UK, as well as prospects for future evidence-based guideline developments. It was not the remit of this roundtable meeting to cover PH in children.
This report of the meeting aims to provide UK healthcare professionals, especially cardiology trainees, and secondary care and primary care workers without specialist knowledge in PH, with information to assist in the management of this complex condition. The panel has also given their opinion on occasions – in areas that might not be particularly clear with current guidance or where evidence is changing, for example – to help in the management of this complex condition.
Background
 Pulmonary hypertension (PH) was described in 2009 guidance from the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) as a haemodynamic and pathophysiological state that can be found in multiple clinical conditions.1 The guidance classified PH into five clinical groups:
Pulmonary hypertension (PH) was described in 2009 guidance from the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) as a haemodynamic and pathophysiological state that can be found in multiple clinical conditions.1 The guidance classified PH into five clinical groups:
1. Pulmonary arterial hypertension (PAH)
1’. Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis
2. PH due to left heart disease
3. PH due to lung disease and/or hypoxia
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. PH with unclear and/or multifactorial mechanisms.
The clinical area of PH is in a process of rapid change at the present time. The 5th World Symposium on PH updated its guidance on PH in 20132 and new guidelines are due from the ESC and ERS in August 2015.
The diagnosis of PH is difficult. There is no singular diagnostic algorithm and misdiagnosis is not uncommon. There are also rapid changes in the evidence base for treatments. Thus, patients previously thought of as untreatable now have a much better prognosis and it is important that they are referred for appropriate treatment and management. In this supplement, we describe some of the more recent developments in PH in order to assist cardiologists when confronted with this complex condition.
Classification
PH was once classified into two categories: primary PH or secondary PH according to the absence or presence of identified causes or risk factors, respectively.3 In 1998, a clinical classification of PH was established categorising PH according to groups that share similar pathological and haemodynamic characteristics and therapeutic approaches.4
Subsequently, amendments have been made to these groupings as knowledge about these conditions grows. Most recently, in 2013, the 5th World Symposium on PH made some minor amendments to this grouping, and the current classification of PH is shown in table 1.2

Within the classification of PH it should always be remembered that PH in many patients has multiple causes. In this situation the most important cause must be identified as the target for treatment while considering any contraindications resulting from other causes. The types of PH that require specialist treatment are group 1 pulmonary arterial hypertension (PAH) and group 4 chronic thromboembolic pulmonary hypertension (CTEPH).
Figure 1 shows how PH is highly heterogenous with some conditions having defined treatments (groups 1, 1’ and 4) and those where there is no evidence for specific pulmonary vasodilator therapy (groups 2, 3 and 5).
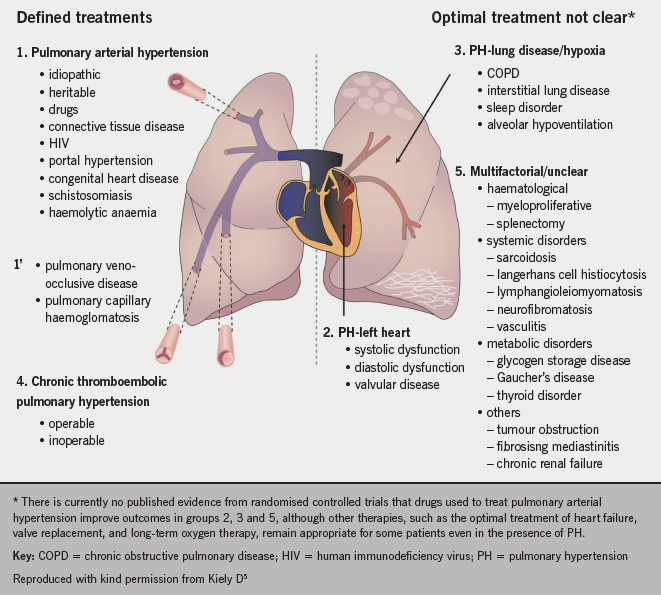
Panel opinion: the classification of PH can be confusing since the terminology used is more suited to specialists in the field than generalists. The classification system does not make it clear who needs to be referred to a pulmonary hypertension centre, as well as conversely, who should not be referred. In particular, the panel considered that the definition of idiopathic pulmonary arterial hypertension (PAH) possibly needs revision to describe the typical severe disease seen in young people as opposed to an atypical form associated with multiple comorbidities more commonly seen in older people.
Epidemiology of PAH
Data from 2012−13 estimate the number of patients with PH cared for in PH specialist centres in Great Britian as being 124 per million of the population, with a greater prevalence in women than men (ratio 1.8:1). The most common causes of diagnosed PH in these specialist centres are PAH (44.9% of patients) and CTEPH (19.2 % of patients).6 Figure 2 shows the increasing incidence of the condition.
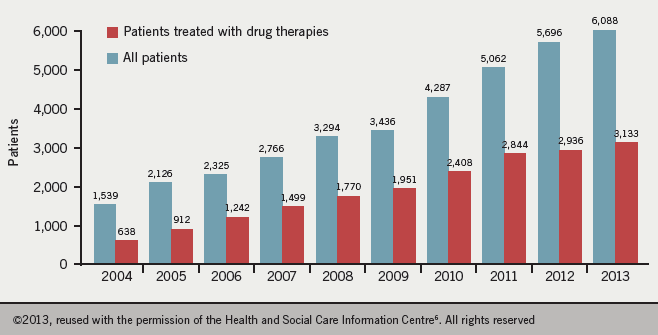
Hypertension Service (on 31st March each year)
But estimating the true prevalence is difficult. It is important to note that many patients with PH are never referred or seen. Epidemiology is different in the general (‘real world’) PH population where group 2 PH dominates. An Australian observational study carried out on all residents referred to a Western Australian unit for transthoracic echocardiography found 326 cases of PH per 100,000 inhabitants, with group 2 PH being the most common with a prevalence of 250 cases per 100,000.7
Studies looking at idiopathic PAH, for example, also reflect how difficult it is to measure prevalence. Idiopathic PAH in the UK and Ireland was estimated at 6.6 per million with an incidence of 1.1 per million per annum between 2001 to 2009,8 whereas ‘real world’ data from a UK national audit from 2009 to 2012 estimated the prevalence at 12–18 per million, although the incidence figure was the same.6 The difference in prevalence can be explained by different criteria used to classify PH. In the national audit all cases were included, whereas Ling et al. excluded patients with any lung disease on computed tomography (CT). It is difficult to exclude patients with heart disease as it is a common finding in older patients with idiopathic PAH. The average age of the cohorts was also different; 50 years in Ling et al. and 66 years in the national audit.
Genetic screening
PH has been linked to a mutation in the gene encoding for BMPR-2: 11–40% of patients with sporadic idiopathic PAH and 70% of patients with a family history of PAH carry such a mutation.5 Such mutations are autosomal dominant, but with a reduced penetrance, such that only 20% of carriers exhibit the disease. There are also epigenetic factors that can increase susceptibility to PAH.9 At present, there are not enough data regarding genetic screening to establish whether it provides benefit, although research is ongoing. However, screening is often available in specialist centres for those who request it.
Role of a specialist centre and referral criteria
Panel opinion: PH is best managed at specialist centres. These can provide patients with support as well as services such as screening, family planning, general surgery and provision of on-call assistance. They can ensure appropriate prescribing and provide a focus for research, which is critical in this poorly understood condition. A map showing specialist centres in the UK is shown in figure 3.
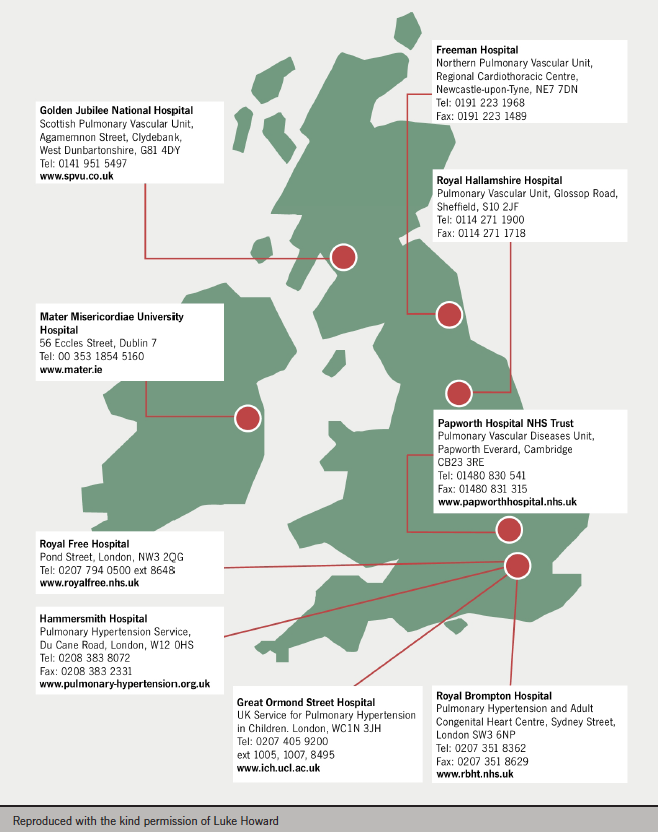
Referrals to specialist centres tend to come from secondary and tertiary care. GP referrals are unusual unless the GP has experienced other patients with PH. The current UK referral criteria are:
- pulmonary hypertension of uncertain cause
- pulmonary arterial hypertension (PAH)
- chronic thrombotic or embolic disease
- miscellaneous causes
- heart and lung disease:- severe pulmonary hypertension (pulmonary artery systolic pressure [PASP] >60 mmHg)- with other PAH-associated disease.
Panel opinion: current UK referral criteria are vague and can lead to inappropriate referrals. It is hoped they will be improved by the review being undertaken by the Clinical Reference Group (CRG) in England. The panel also felt that clarification was required in heart and lung disease, where it is important to look for patients with PH and disproportionate symptoms i.e. symptoms that do not fit the clinically evident disease.
Diagnosis
Symptoms of PH are non-specific and the signs are often subtle. There are two complementary approaches to discovering cases of PH. First, the systematic evaluation of the breathless patients and, second, looking for the diagnosis in high-risk groups.
It is important to be aware of:
- the varied clinical presentation of PH- indicators can include breathlessness, fatigue, chest tightness, syncope, while haemoptysis and ankle oedema/ascites are features that may be seen in later disease.- examination may reveal increased jugular venous pressure, loud P2 and parasternal heave (PSH) may be seen on imaging.
- populations with a high prevalence of treatable PAH1 including- those with connective tissue disease (up to 19% of those with systemic sclerosis)- congenital heart disease (approximately 5–10% mainly in the form of Eisenmenger Syndrome)- portal hypertension (2–6%) – HIV infection (0.5%)- pulmonary thromboembolism (0.5–5%)- those with a family history of PAH.
- respiratory and cardiac diseases complicated by PH including – chronic obstructive pulmonary disease (COPD), interstitial lung disease (ILD) and alveolar hypoventilation – valvular heart disease, systolic and diastolic dysfunction.
The fundamental criterion for diagnosis of PH is the mean pulmonary artery pressure (PAP) measurement. Cardiac catheterisation is the gold-standard for measuring PAP and diagnosis is confirmed with a PAP ≥25 mmHg. Cardiac catheterisation, however, generally does not provide information on the cause of the PH. This procedure is covered in more detail later in the supplement. It should be noted that echocardiography is not an accurate method for measuring PAP.
In the presence of respiratory and cardiac disease, PH is a common finding that can have implications for prognosis. Generally, however, the PH is mild and does not warrant referral or treatment. In cases where the PH is significant and ‘out of keeping’ with the comorbid conditions, referral to a specialist centre is advised.
Panel opinion: the diagnosis of PH is a good example of where the practice of medicine is as much an art as a science.
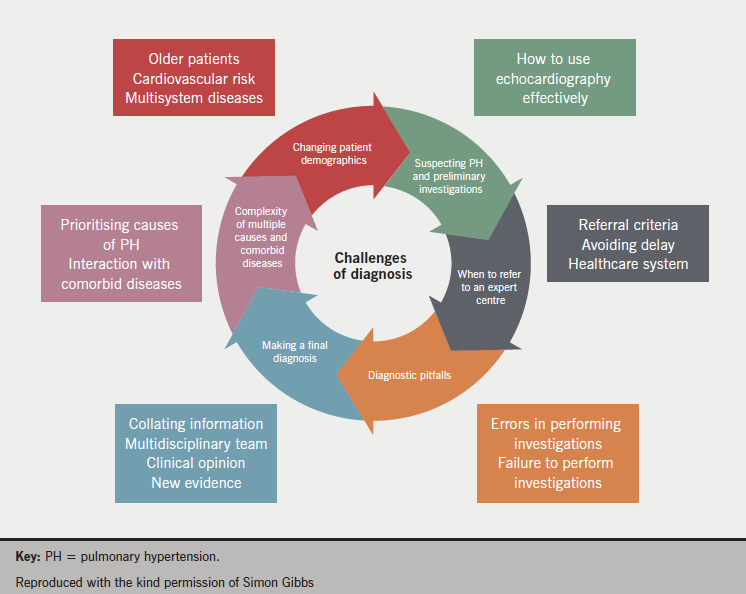
Challenges of diagnosis
The characteristics of the various PH populations are changing as the demographics of the general population change. Increasing age and body mass index (BMI) represent a challenge for the diagnosis and treatment of PH. Older patients often have different clinical, physiological and haemodynamic characteristics and more comorbidities. In obese patients, in addition to practical issues concerning conducting cardiac catheterisation, increased PAP is also a common finding. High BMI is often associated with obstructive sleep apnoea and hypoventilation, with abnormal lung function a common finding. Heart disease is also commonly present in this population group.
In general, older people take longer to get a diagnosis, probably because breathlessness is common in older people. The characteristics of idiopathic PAH, for example, are also different in older populations compared with younger people. Key differences in older people include:8,10
- worse survival
- less good response to PAH drug therapy
- higher pulmonary artery wedge pressure (PAWP)
- lower PAP and peripheral vascular resistance (PVR) for a given cardiac index
- prolonged delay in diagnosis
- likely to present with severe symptoms
- shorter six-minute walk distance (6MWD).
The survival of patients varies considerably with the type of PH.11 Misclassification of patients can provide erroneous prognostic data, leading to inappropriate counselling of patients, so it is important to fully investigate the cause of PH and accurately phenotype it.
PAH
Looking at PAH, there are many challenges involved in navigating comorbidity maze. First, it is important to diagnose the right form of PH:
- PAH versus heart failure with preserved ejection fraction (HFpEF)
- PAH versus severe PH in respiratory disease
- PAH versus CTEPH.
Then the cause of the PAH should be identified, is it idiopathic? Idiopathic PAH is a diagnosis of exclusion, which is challenging in the presence of comorbid diseases. The value and limitations of different diagnostic tools should be recognised and it should also be appreciated that patients can exhibit more than one disease phenotype. The challenges of diagnosis are listed in
figure 4.
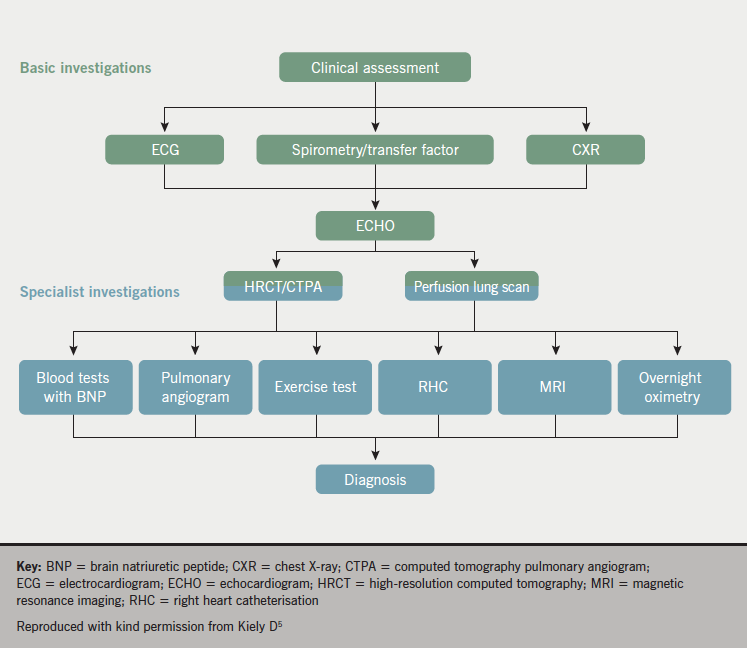
Investigations
Panel opinion: in the diagnosis of PH, a narrative should be built where each investigation provides a piece of information that needs to be put together with all the other investigations to provide an overall picture.
Multiple tests are required in order to establish a diagnosis of PH since no single test can provide a diagnosis. An investigative approach to diagnosing PH is provided in figure 5. Chest X-ray (CXR), electrocardiogram (ECG), echocardiography and computed tomography pulmonary angiography (CTPA) are helpful in the initial assessment of unexplained PH.
CXR, ECG and PFTs
CXR and ECG may suggest PH in a breathless patient. An abnormal CXR or ECG is seen in up to 90% of patients with idiopathic PAH, for example.12
Pulmonary function tests (PFTs) are essential to the diagnosis and evaluation
of PH.
Spirometry will often demonstrate mild restriction in forced vital capacity (FVC) and reduced gas transfer (TLCO) values.13 It should be noted that a TLCO <50% of predicted should raise suspicion of systemic sclerosis or co-existing lung disease or pulmonary veno-occlusive disease (PVOD).13
Perfusion lung scanning has a high sensitivity for CTEPH,14 and is recommended to exclude CTEPH.1
While a valuable investigative tool in this regard, availability may be limited.
Echocardiography
If PH is suspected, echocardiography should not be delayed while waiting for an ECG, CXR or respiratory investigations. Appearance on echocardiography will include right ventricular (RV) and right atrial (RA) dilation. Tricuspid annular plane systolic excursion (TAPSE) is usually reduced and paradoxical septal motion may be observed. Echocardiography should also include a valvular assessment and estimation of left atrial (LA) size, as well as estimation of systolic PAP (sPAP) where: sPAP = 4V2 + RA.15
Echocardiography is less accurate in respiratory disease, since an estimation of systolic PAP is not possible in about three out of five patients but it is frequently overestimated when it is possible.16 Therefore, when evaluating respiratory patients, PH should be considered unlikely if there are no RV findings (dilatation, hypertrophy, systolic dysfunction) or the estimated pulmonary artery systolic pressure (est PASP) is <45 mmHg.
CTPA
Computed tomography pulmonary angiography (CTPA) has the potential to identify causes of PH and establish the severity. It is important to assess:
- vessels
- heart
- mediastinum
- lung parenchyma.
Pulmonary artery size is not specific and an enlarged pulmonary artery on CT does not necessarily indicate PH. Other tests should be performed prior to referral to a specialist centre.
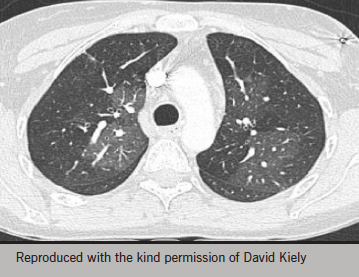
Geographic mosaicism is characteristic of CTEPH (figure 6)17,18 but may also be seen in small airways disease and severe PAH including PVOD.
Ground-glass opacity on CT refers to areas of increased attenuation that do not obscure vessels17 and in Eisenmenger’s syndrome they correlate with dilated capillary networks.19 They also correlate with risk of treatment failure,20 and are common in all forms of PAH.21
MRI
The role of magnetic resonance imaging (MRI) in PH is evolving. At present, there is no evidence to support the role of MRI in the initial diagnosis of PAH but it can provide useful information to assist in diagnosis. Potential roles for this imaging technique include:
- diagnosis of PH
- identifying the cause of PH
- identification of patients with CTEPH suitable for surgery
- assessing disease severity and response to therapy
- estimation of pulmonary haemodynamics.
Catheterisation and right heart catheter values
As we have already learned, cardiac catheterisation is the gold standard investigation for the diagnosis of PH.
It is a procedure which should be carried out in expert, high-volume centres as it needs care and attention to detail. Normal resting mean PAP is 8–20 mmHg; a mean resting PAP of ≥25 mmHg is definitive for PH.22
Further measurements taken during the procedure, such as estimating pulmonary artery wedge pressure (PAWP) and the cardiac output, can provide additional information. For example:
- if PAWP is >15 mmHg at end expiration, the PH is post-capillary and likely to be due to left heart disease (check saturations with balloon up), and there is usually a significant increase in right arterial pressure
- if PAWP is <15 mmHg with normal or low cardiac output, then it is likely to be a pre-capillary cause of PH, e.g. PAH, PH from respiratory causes, CTEPH, or multi-factorial in origin
- if PAWP is <15 mmHg with an increased cardiac output, this can indicate left to right shunt, but can also be seen in portopulmonary hypertension and other causes of increased cardiac output such as hyperthyroidism. These patients should go on to have further saturation measurements.
Since the World Symposium, it has been recommended that the diagnosis of PAH should also include a pulmonary vascular resistance (PVR) >3 Wood units in addition to a mean PAP ≥25 mmHg, and a PAWP ≤15 mmHg. The inclusion of PVR in the haemodynamic definition of PAH is intended to avoid the misdiagnosis of circulatory failure from a high output state as PAH.
Pulmonary vascular resistance (PVR) is a measure of resistance in the pulmonary circulation: PVR (Wood units) = (mean PAP – PAOP)/cardiac output (CO).
PVR is usually 1–2 Wood Units or 80–160 dynes. It is elevated in PAH and must be ≥3 Wood units. It is usually 10+ in idiopathic PAH but lower in PAH associated with connective tissue disease (PAH-CTD) and higher in PAH associated with congenital heart disease (PAH-CHD). In patients with left-to-right shunts it can be normal or mildly elevated.
Panel opinion: cardiac catheterisation is best carried out in expert centres. The panel expressed concern at the level of training doctors now receive in haemodynamics and that, if the right level of care is not taken, the values obtained by catheterisation could be as inaccurate as those obtained by echocardiography.
Other investigations
Overnight oximetry may suggest obstructive sleep apnoea (OSA)/hypoventilation.11 There are a number of investigations used in clinical practice that lack robust evidence in PH. This is not to say that they should not be used clinically. These include cardiopulmonary exercise testing and exercise echocardiography, as well as fluid loading and exercise during cardiac catheterisation.
PH summary
- Pulmonary hypertension has many causes
- Diagnosing PAH requires a systematic approach to the breathless patient and screening of high-risk groups
- Navigating the PAH ‘comorbidity maze’ requires careful history and examination and multi-modality assessment
- Accurate classification can be challenging with many potential pitfalls
- Specialist PH centres provide an environment where patients can be evaluated systematically, as well as offering support and treatment for patients
Treatment
As we have seen in figure 1, there are defined treatments for patients with PAH (group 1) and CTEPH (group 4). The treatment of PAH is covered in this section and CTEPH is covered later in the supplement.
In clinical practice, the following are used to determine the response to therapy and prognosis in patients with PAH:
- functional class
- echocardiography/cardiac MRI
- haemodynamics
- 6MWD
- cardiopulmonary exercise testing
- B-type natriuretic peptide level.
Algorithms for the treatment of PAH vary across the globe. In England, the National Health Service Executive (NHSE) drug policy must be followed. The use of sequential combination therapy is now standard in PAH although some patients do not progress beyond monotherapy. During the round table meeting, changes in treatment guidelines and the evidence for progressing to increased use of combination therapy were discussed.
Recent guidelines
In 2013, the World Symposium on PH updated the treatment algorithm for PAH (figure 7).23 Of note was the upgrading of supervised exercise training due to recent randomised controlled trials (RCTs). This training needs to be supervised closely and should only be offered to patients receiving optimal treatment. Due to the PH, there is a high risk of syncope and other complications in even the most experienced hands. For this and other reasons, standard cardiac rehabilitation programmes are unlikely to be suitable for patients with PH.

General measures and supportive therapy are important to the overall management of the patient. As well as supervised exercise training, patients should receive psychosocial support, influenza and pneumococcal immunisation and be advised to avoid strenuous physical activity and pregnancy. Supportive therapy should include oral anticoagulants, diuretics, digoxin and oxygen, as appropriate.23
There are also considerations in light of new combination therapy data. Sequential combination therapy is now one of the initial therapy recommendations. Sequential combination therapy continues to be recommended in the event of unsatisfactory response to monotherapy. The evidence base is discussed later.
Treatment pathways
There are three key vasomotor pathways targeted by current and emerging therapies in PAH, shown in figure 8.
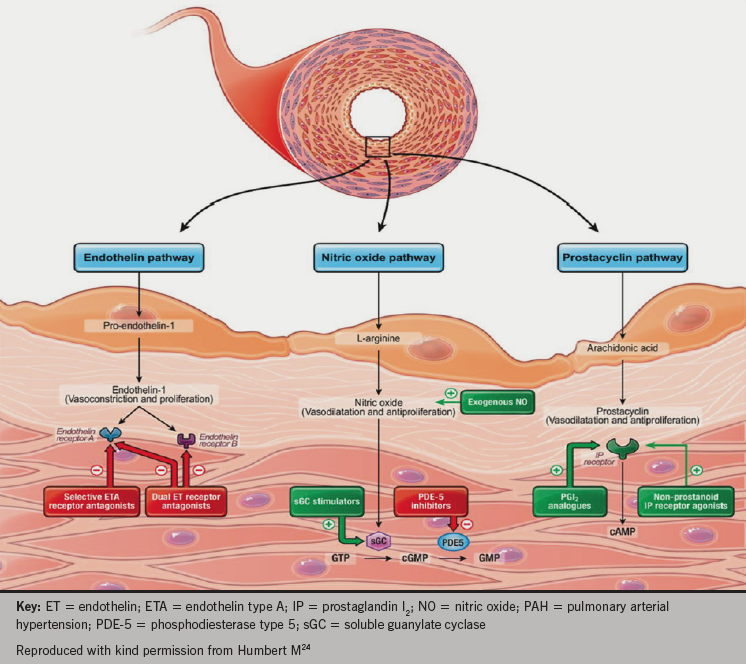
therapies in PAH
- The prostacyclin pathway: prostanoids mimic the effects of prostacyclin (which is deficient in patients with PH) and produce potent vasodilatation. Prostanoids include epoprostenol, iloprost and treprostinil. Selexipag (currently submitted for regulatory approval in Europe and the USA) is a selective prostacyclin IP receptor agonist.
- The endothelin pathway: endothelin receptor antagonists counter the vasoconstricting effects of endothelin-1, which is over produced in the lungs of patients with PAH and may mediate remodeling effects in the vasculature. Bosentan and macitentan act at both the A and B type endothelin receptors, while ambrisentan is selective for the endothelin-A receptor.
- The nitric oxide (NO) pathway: production of endogenous NO is reduced in patients with PAH. NO is a vasodilator and a signalling molecule important in the production of the intracellular messenger cyclic guanine monophosphate (cGMP). Phosphodiesterase type-5 inhibitors, which include sildenafil and tadalafil, prevent cGMP degradation, thus promoting the NO/cGMP pathway with consequent vasodilatation. More recently, a newer class of agents has been used in the treatment of PH: soluble guanylate cyclase stimulators. They stimulate the action of cGMP to reduce intracellular calcium in both an NO dependent and independent fashion. Riociguat has been trialled in patients with PAH and CTEPH.25–7
Clinical studies
Over the last 15 years there have been over 30 RCTs of PAH therapies. Historically, in most studies, 6MWD has been used as the primary end point. This end point allows realistic numbers to be included in a trial and a shorter trial duration. While exercise capacity is important in very sick, young patients, change in 6MWD is not a surrogate of clinical outcome.
Historically, time to clinical worsening (TTCW) has sometimes been included as a secondary end point, but studies using this were often not powered to be able to detect a significant difference in this end point. More recently, trials have included a combined morbidity/mortality primary end point e.g. SERAPHIN (Study with an Endothelin Receptor Antagonist in Pulmonary arterial Hypertension to Improve cliNical outcome),28 AMBITION (A Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects With Pulmonary Arterial Hypertension),29 GRIPHON (The Prostacyclin [PGI2] Receptor Agonist In Pulmonary Arterial Hypertension)30and COMPASS 2 (Effects of the Combination of Bosentan and Sildenafil Versus Sildenafil Monotherapy on Pulmonary Arterial Hypertension).31 It is important to note that none of these studies were actually powered to show a mortality benefit.
Combination treatment
Many trials have used sequential combination therapy in the treatment of PH but they have not been powered to look at the benefit of combination therapy versus monotherapy. Several meta-analyses have been conducted on these trials with mixed results: while an improvement in 6MWD with combination therapy is observed, improvements in mortality are not apparent, although it should be noted these studies were not powered to investigate mortality.32–4
SERAPHIN looked at macitentan versus placebo in treatment naïve patients or on top of stable background therapy. In the group of patients who were already on background therapy, the morbidity/mortality driven end point was improved by 38% with the addition of macitentan to background therapy compared to those patients who received placebo plus background therapy. Much of this benefit was due to a reduction in hospital admissions. The combination treatment was generally well tolerated.28
In the GRIPHON study, the addition of selexipag to a stable dose of either an endothelin-receptor antagonist (ERA) or a PDE-5 inhibitor produced a 39% reduction in the risk of a morbidity/mortality event versus placebo over up to 4.3 years.30
In comparison, in COMPASS-2, the sequential addition of bosentan to sildenafil therapy failed to demonstrate a significant reduction in time to first morbidity/mortality event, although there was a 17% reduction versus placebo (p=0.25). There was also improvement in 6MWD at 16 weeks.31
Panel opinion: this result stresses the importance of non-generalisation of specific combinations of agents.
Importantly, AMBITION looked at initiation with combination therapy, rather than sequential add-on, comparing ambrisentan and tadalafil to tadalafil or ambrisentan monotherapy. The combination arm was superior to the pooled monotherapy arms, as well as the individual monotherapies for the primary end point of time to clinical failure. There was a 50% decrease in clinical failure events with the combination treatment compared with monotherapy, and the greatest benefit was gained in those with lower WHO functional class.29
Finally, there has been a small, open label, pilot study of triple therapy (intravenous epoprostenol, bosentan and sildenafil) involving 19 subjects that had promising results. In the 18 patients still receiving triple therapy after three years, all had sustained clinical and haemodynamic improvement. Survival in this trial at three years was 100%, which compares to a predicted survival of 49%.35
Panel opinion: while noting this study is small and not able to be generalised, the panel felt its findings add weight to the argument that aggressive early treatment targeting multiple pathways has the potential to improve outcomes (enhanced systemic vasodilation).
Considerations in combination therapy
As mentioned above, not all combinations of PAH treatments are the same. As more RCTs are conducted with appropriate end points, the most effective combinations should become clearer. The relative difference of the timing of the combination treatment should also be clarified and the importance, or not, of initiating combination treatment early will be established. The impact of patient age and physical status on the efficacy of product combinations, as well as cost-effectiveness, also remain to be established. It is already clear that some combinations should always be avoided such as riociguat and PDE-5 inhibitors.23
Transplantation
Advanced therapies appear to have delayed referrals for bilateral sequential lung transplantation but this should remain an option in patients in WHO functional class IV and those who remain in WHO functional class III despite combination therapy. The increased use of extracorporeal lung support may influence the outcome of transplantation.
Transplantation should be considered a priority for pulmonary veno-occlusive disease (PVOD)/pulmonary capillary hemangiomatosis (PCH). Some conditions, such as PAH related to connective tissue disease (PAH-CTD), are a relative contraindication.
Despite this, transplant outcomes are improving with survival figures of 52–75% at five years, and 45–66% at 10 years.23 Transplantation is not common – worldwide 122 transplants for PAH were performed in 2012 and 70 in 2013. In the UK, around four to five transplants are performed per year. It is important that patients are not referred too late for transplantation.
Treatment goals
There is currently no validated method for monitoring PAH during follow-up. It is also not clear if all treatment goals are appropriate for all the different patient subgroups.
A combination of measures should be followed in order to fully assess patient response to treatment. Assessments currently used to assess patients include:
- WHO functional class
- 6MWD
- quality of life
- biomarkers
- echocardiography
- cardiac MRI
- invasive haemodynamics
- cardiopulmonary exercise testing (CPET).
Table 2 shows which parameters are better predictors of prognosis.
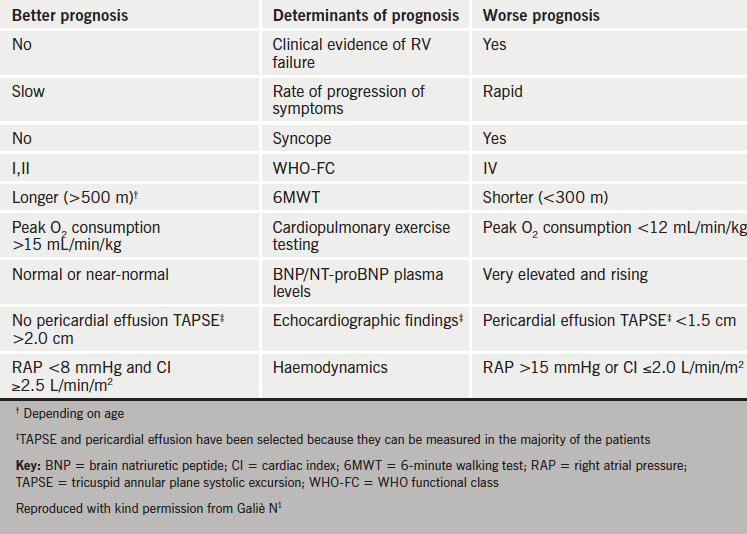
prognosis in PAH
Panel opinion: it is essential that all patients with PH should be actively managed and followed-up at least every three to six months. Women of child-bearing age should be given contraceptive advice.
WHO Functional Class
Baseline WHO functional class is predictive of survival and also the change in functional class can show the benefit of treatment. While this is a simple and reproducible measure, inter-observer reliability is poor.
6MWD
The six-minute walk distance is a simple, cheap, reproducible and well-tolerated measure of exercise capacity. Baseline 6MWD correlates to haemodynamics and survival. Furthermore, threshold values after treatment predict survival. Worsening 6MWD is strongly and significantly associated with poor prognosis.36 It is used in many clinical trials but it does have some limitations. There is a ceiling effect. Only 22% of the mortality benefit associated with treatment can be predicted from change in 6MWD,37 while the minimal required change in 6MWD for a measurable improvement in quality of life is around 33 m.38 Thus there are some problems with using 6MWD to assess outcome in combination treatment studies.
CPET
CPET provides an integrative approach to assessing cardiac function, gas exchange and muscular physiology. Peak oxygen consumption has been shown to be predictive of survival in three studies (10.4 ml/min/kg, 11.5 ml/kg/min and 13.2 ml/min/kg, below which mortality increased). In terms of goals for therapy, it is suggested that <10 ml/min/kg indicates a poor prognosis, whereas >15 ml/kg/min indicates a better prognosis. Ventilatory equivalents for CO2 (EqCO2) <45 should be used as a treatment goal, and >55 as a warning sign.
Biomarkers
Brain natriuretic peptide (BNP) and N-terminal pro-BNP (NT-proBNP) are age and sex dependent. BNP levels generally parallel haemodynamic and functional data. Thus, a treatment goal is ‘normalisation’ of BNP with therapy. A follow-up NT-proBNP <1,800 pg/ml has been shown to indicate a better prognosis in the current treatment era.39 Changes in NT-proBNP have also been shown to be highly predictive of outcome in sclerodoma-associated PAH.40
Echocardiography
Current recommendations are to achieve normal/near normal RV size and function. However, there are problems regarding agreement on the best methods to measure and report these. The Tei index, TAPSE, eccentricity index, RA area, RV enlargement, and presence of pericardial effusion have been shown to be predictive of survival.
Cardiac MRI
Cardiac MRI is the gold standard for assessing RV structure and volumes. A right ventricular end-diastolic volume index (RVEDVI) <84 ml/m2, left ventricular end-diastolic volume index (LVEDVI) >40 ml/m2 and stroke volume index (SVI) >25 ml/m2 are associated with better survival in patients with idiopathic PAH.41 Data are currently limited with follow-up.
Haemodynamic parameters
Numerous studies have indicated that right atrial pressure (RAP), cardiac index (CI) and mixed venous oxygen saturation (SvO2) are good indicators of prognosis.42 Treatment goals should be RAP <8 mmHg, CI >2.5 L/min/m2 and SvO2 >65%. Measurement of these parameters is invasive, generally at rest and occurs at one point in time. At present there is no consensus on the utility of exercise right heart catheterisation.
PAH treatment summary
- Prognosis for patients with PAH is improving
- A combination of parameters is needed to establish whether treatment goals are being met
- Majority of patients will not have a satisfactory outcome on monotherapy
- Patients need active management and advancement in therapy
CTEPH
Chronic thromboembolic pulmonary hypertension (CTEPH) is a distinct cause of PH due to failure of fibrinolysis from pulmonary thromboembolism but with the same haemodynamic criteria as PAH (defined by mean PAP ≥25 mmHg).1
CTEPH has two components:
- The organised thromboembolic material that is causing the initial obstruction (thrombembolic obstruction may be in the proximal or distal pulmonary artery).
- A secondary vascular phenomenon, which may explain the ‘honeymoon’ period with patients apparently recovering substantially from the initial pulmonary embolic event only to develop progressive symptomatic limitation months or years later. In this setting, surgery to remove occlusive material from as few as six of the 20 segmental pulmonary vessels is often sufficient to normalise pulmonary pressures.
CTEPH is relatively rare, potentially fatal, but also potentially curable with pulmonary endarterectomy.43,44
Epidemiology
CTEPH is thought to develop following a pulmonary embolism (PE) that fails to resolve in between 0.57% and 9.1% of patients.45–9The largest epidemiological study followed 866 patients with PE. They observed a total cumulative incidence of CTEPH of 0.57%.50 Some 25–63% of patients with CTEPH do not have a previously documented venous thromboembolism (VTE).51–4 Therefore, the true incidence of CTEPH is likely to be underestimated by studies that only follow patients after a PE. This highlights that all patients with PH should be assessed for CTEPH even in the absence of a prior PE.
In the UK, the prevalence of diagnosed CTEPH is 19.2 per million, equating to 1,169 cases in 2012–2013.6 In 2012–2013, of the 2,146 new referrals to pulmonary hypertension centres, in those in whom a final diagnosis was made, CTEPH accounted for 12.8% (222) of these referrals. It has also been observed that the incidence of CTEPH is higher in areas in close proximity to a PH centre. Thus, it is possible that the true incidence and prevalence are actually much greater throughout the whole of the UK.
Diagnosis
Panel opinion: multi-modality imaging should become the gold standard for diagnosing CTEPH.
Pulmonary angiography is regarded as the gold-standard diagnostic tool in the work up of CTEPH. This should be carried out by centres of excellence to ensure that results are interpreted correctly. Imaging is a rapidly evolving field and often a combination of techniques is required to confirm a diagnosis of CTEPH. Again, specialist centres can have access to more advanced imaging techniques with the personnel best placed to interpret them.
A ventilation/perfusion (V/Q) lung scan is sensitive for CTEPH but not specific for the condition. Unfortunately this investigation is not available in many centres, which may result in many cases being missed. The more widely available CTPA is specific for CTEPH, but not sensitive. Sensitivity can be increased when performed by an experienced operator. It is important that scans are performed in 1 mm slices, read by experts and that mosaic perfusion patterns are looked for (figure 6).
Treatment
The prognosis of CTEPH has improved considerably in the last 40 years, particularly in those patients who are operable and undergo surgery (pulmonary endarterectomy), the treatment of choice in this condition. Surgery works well because the initiating cause is removed, which appears to allow the vascular response to improve in those patients. Mortality rates for surgery have improved considerably and are lower in larger centres performing larger numbers of procedures than in small volume centres. In the UK, Papworth Hospital is the single, national centre commissioned to provide pulmonary endarterectomy and its mortality rate in 2012−13 was a very low 2% making it one of the best centres in the world for this procedure. There is, however, considerable morbidity associated with the procedure and almost half of patients (49%) experience complications.
Around a third of patients, however, are not suitable for surgery and about a fifth are likely to have residual PH after the operation. Alternatives to surgery are actively being sought but surgery is currently regarded as the definitive treatment. Recent National Audit figures have shown that operated CTEPH patients have a much better three-year survival rate (90%) than CTEPH patients who are not operated (70%).6
Drug treatment is reserved for patients who are unable to undergo surgery or have failed post-surgery.
The BENEFiT (Bosentan Effects in Inoperable Forms of Chronic Thromboembolic Pulmonary Hypertension) study was a RCT in 157 patients with inoperable CTEPH, or residual or recurrent PH following pulmonary endarterectomy, with the co-primary end points of change in PVR and 6MWD. Bosentan demonstrated a significant effect over placebo for PVR (–24.1% of baseline, p<0.0001) although no improvement was observed in exercise capacity.55 Bosentan is currently not licensed for use in CTEPH in the UK.
Riociguat is the first proven therapy to improve exercise capacity (6MWD) and quality of life/symptoms in this population. CHEST-1 (Chronic Thromboembolic Pulmonary Hypertension Soluble Guanylate Cyclase-Stimulator Trial 1) investigated riociguat versus placebo in 261 patients with inoperable CTEPH or persistent or recurrent PH after pulmonary endarterectomy using 6MWD as the primary end point. By week 16, a significant increase in 6MWD was demonstrated for riociguat versus placebo (39 m vs. –6 m, p<0.001). Significant improvement in PVR (p<0.001) and WHO functional class (p=0.003) was also observed.26 Long-term follow-up of patients has shown that riociguat can provide sustained benefits in exercise and functional capacity for up to one year.56
Balloon angioplasty is a non-surgical option that is currently being explored as a potential future treatment for patients with CTEPH who cannot undergo surgery. Further improvements in imaging techniques will help to develop this option.
As the technique of ballon pulmonary angioplasty develops, we need a greater understanding of precise lesion types and their response to balloon dilation. Furthermore, we need standardisation of terminology to share knowledge and construct appropriate studies. The following classification has been proposed by Sugiyama et al.:57
- type 1 a web at a bifurcation from segmental into subsegmental arteries
- type 2 a web with distal slits
- type 3 only a slit in the subsegmental artery
- type 4 a complete occlusion.
Currently the resolution required to differentiate these lesion types is available with cone-beam CT, but the clinical usefulness of this technique is limited by the small field of view and motion artefacts. It has recently been observed that ECG-gated 320-row CT can provide similar detail.
MR pulmonary angiogram could also be a promising modality but this has not yet been formally assessed.
CTEPH summary
- CTEPH is relatively common and probably under-diagnosed
- Failure of normal fibrinolysis is the key initiating process but there is an additional proliferative vasculopathy
- Pulmonary endarterectomy surgery is the treatment of choice – but this is a big operation with a high morbidity burden
- Drug therapy and balloon pulmonary angioplasty (BPA) are likely to play an increasing role in the near future
- Improved CT resolution is likely to improve our ability to diagnose and optimise management
Key messages
- Outcomes in PAH and CTEPH are improving and it is a treatable condition for many patients
- Morbidity and mortality remains considerable
- Management of patients with PH is complex and should be carried out by specialist centres with regular follow-up
- Specialist centres provide expert diagnostics, treatment and support for patients
References
1. Galiè N, Hoeper MM, Humbert M et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009;30:2493–537. http://dx.doi.org/10.1093/eurheartj/ehp297
2. Simonneau G, Gatzoulis MA, Adatia I et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013;62(suppl D):D34–D41. http://dx.doi.org/10.1016/j.jacc.2013.10.029
3. Hatano S, Strasser T, editors. Primary Pulmonary Hypertension. Report on a WHO Meeting. Geneva: World Health Organization, 1975:7–45.
4. Simonneau G, Galiè N, Rubin LJ et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43(suppl):5S–12S. http://dx.doi.org/10.1016/j.jacc.2004.02.037
5. Kiely DG, Elliot CA, Sabroe I, Condliffe R. Pulmonary hypertension: diagnosis and management. BMJ 2013;346:f2028. http://dx.doi.org/10.1136/bmj.f2028
6. Health and Social Care Information Centre. National Pulmonary Hypertension Audit –2013. London: HSCIC, 2014. Available from: http://www.hscic.gov.uk/catalogue/PUB13318/nati-pulm-hype-audi-2013-rep.pdf
7. Strange G, Playford D, Stewart S, Deague JA, Nelson H, Kent A, Gabbay E. Pulmonary hypertension: prevalence and mortality in the Armadale echocardiography cohort. Heart 2012;98:1805−11. http://dx.doi.org/10.1136/heartjnl-2012-301992
8. Ling Y, Johnson MK, Kiely DG et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension. Results from the Pulmonary Hypertension Registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012;186:790–6. http://dx.doi.org/10.1164/rccm.201203-0383OC
9. Tuder RM, Archer SL, Dorfmuller P et al. Relevant issues in the pathology and pathobiology of pulmonary h ypertension. J Am Coll Cardiol 2013;62(25 Suppl):D4–D12. http://dx.doi.org/10.1016/j.jacc.2013.10025
10. Hoeper MM, Huscher D, Ghofrani HA et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013;168:871–80. http://dx.doi.org/10.1016/j.ijcard.2012.10.026
11. Hurdman J, Condliffe R, Elliot CA et al. ASPIRE registry: assessing the spectrum of pulmonary hypertension identified at a referral centre. Eur Respir J 2012;39:945–55. http://dx.doi.org/10.1183/09031936.00078411
12. Rich S, Dantzker DR, Ayres SM et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987;107:216–23. http://dx.doi.org/10.7326/0003-4819-107-2-216
13. Sun XG, Hansen JE, Oudiz RJ, Wasserman K. Pulmonary function in primary pulmonary hypertension. J Am Coll Cardiol 2003;41:1028–35. http://dx.doi.org/10.1016/S0735-1097(02)02964-9
14. Tunariu N, Gibbs SJ, Win Z et al. Ventilation-perfusion scintigraphy is more sensitive than multidetector CTPA in detecting chronic thromboembolic pulmonary disease as a treatable cause of pulmonary hypertension. J Nucl Med 2007;48:690–4. http://dx.doi.org/10.2967/jnumed.106.039438
15. Howard LS, Grapsa J, Dawson D et al. Echocardiographic assessment of pulmonary hypertension: standard operating procedure. Eur Resp Rev 2012;21:239–48. http://dx.doi.org/10.1183/09059180.00003912
16. Arcasoy SM, Christie JD, Ferrari VA et al. Echocardiographic assessment of pulmonary hypertension in patients with advanced lung disease. Am J Respir Crit Care Med 2003;167:735–40. http://dx.doi.org/10.1164/rccm.200210-1130OC
17. Sherrick AD, Swensen SJ, Hartman TE. Mosaic pattern of lung attenuation on CT scans: frequency among patients with pulmonary artery hypertension of different causes. Am J Roentgenol 1997;169:79–82. http://dx.doi.org/10.2214/ajr.169.1.9207504
18. Kauczor HU, Schwickert HC, Mayer E, Schweden F, Schild HH, Thelen M. Spiral CT of bronchial arteries in chronic thromboembolism. J Comput Assist Tomogr 1994;18:855–61. http://dx.doi.org/10.1097/00004728-199411000-00002
19. Sheehan R, Perloff JK, Fishbein MC, Gjertson D, Aberle DR. Pulmonary neovascularity: a distinctive radiographic finding in Eisenmenger syndrome. Circulation 2005;112:2778–85. http://dx.doi.org/10.1161/CIRCULATIONAHA.104.509869
20. Resten A, Maitre S, Sumbert M et al. Pulmonary arterial hypertension: thin-section CT predictors of epoprostenol therapy failure. Radiology 2002;222:782–8. http://dx.doi.org/10.1148/radiol.2223010668
21. Rajaram S, Swift AJ, Condliffe R et al. CT features of pulmonary arterial hypertension and its major subtypes: a systematic CT evaluation of 292 patients from the ASPIRE registry. Thorax 2014; pii. http://dx.doi.org/10.1136/ thoraxjnl-2014-206088
22. Badesch DB, Champion CC, Sanchez MAG et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009;54 (Suppl S):S55−S66. http://dx.doi.org/10.1016/j.jacc.2009.04.011
23. Galie N, Corris PA, Frost A et al. Updated treatment algorithm of pulmonary arterial hypertension. J Am Coll Cardiol 2013;62(suppl D): D60–D72. http://dx.doi.org/10.1016/j.jacc.2013.10.031
24. Humbert M, Lau EMT, Montani D, Jaïs X, Sitbon O, Simonneau G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014;130:2189–208. http://dx.doi.org/10.1161/CIRCULATIONAHA.114.006974
25. Ghofrani HA, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013;369:330−40. http://dx.doi.org/10.1056/NEJMoa1209655
26. Ghofrani HA, D’Armini AM, Grimminger F et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med 2013;369:319–29. http://dx.doi.org/10.1056/NEJMoa1209657
27. Rubin LJ, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J 2015 (published online 22nd January 2015) http://dx.doi.org/10.1183/09031936.00090614
28. Pulido T, Adzerikho I, Channick RN et al. Macitentan and morbidity and mortality in pulmonary hypertension. N Engl J Med 2013;369:809–18. http://dx.doi.org/10.1056/NEJMoa1213917
29. AMBITION study 2014. First-line combination of ambrisentan and tadalfil reduces risk of clinical failure compared to monotherapy in pulmonary arterial hypertension outcomes study. Abstract 2916. Presented at the European Respiratory Society International Congress, Munich, Germany, 8th September 2014. Available from: http://www.gilead.com/news/press-releases/2014/9/firstline-combination-of-ambrisentan-and-tadalafil-reduces-risk-of-clinical-failure-compared-to-monotherapy-in-pulmonary-arterial-hypertension-outcomes-study
30. McLaughlin VV, Channick R, Chin K et al. Effect of selexipag on morbidity/mortality in pulmonary arterial hypertension: results of the GRIPHON study. J Am Coll Cardiol 2015;65. http://dx.doi.org/10.1016/S0735-1097(15)61538-8
31. McLaughlin V et al. Effect of bosentan and sildenafil combination therapy on morbidity and mortality in pulmonary arterial hypertension (PAH): results from the COMPASS-2 study. Abstract 1992777. Presented CHEST 2014, Austin, Texas USA, October 25th–30th 2014.
32. Fox BD, Shimony A, Langleben D. Meta-analysis of monotherapy versus combination therapy for pulmonary arterial hypertension. Am J Cardiol 2011;108:1177–82. http://dx.doi.org/10.1016/j.amjcard.2011.06.021
33. Coeytaux RR, Schmit KM, Kraft BD et al. Comparative effectiveness and safety of drug therapy for pulmonary arterial hypertension: a systematic review and meta-analysis. Chest 2014;145:1055–63. http://dx.doi.org/10.1378/chest.13-1864
34. Zhu B, Wang L, Sun L, Cao R. Combination therapy improves exercise capacity and reduces risk of clinical worsening in patients with pulmonary arterial hypertension: a meta-analysis. J Cardiovasc Pharmacol 2012;60:342–6. http://dx.doi.org/10.1097/FJC.0b013e318262a793
35. Sitbon O, Jais X, Savale L et al. Upfront triple combination therapy in pulmonary arterial hypertension: a pilot study. Eur Respir J 2014;43:1691–7. http://dx.doi.org/10.1183/09031936.00116313
36. Farber MW, Miller DP, McGoon MD, Frost AE, Denton WW, Benza RL. Predicting outcomes in pulmonary arterial hypertension based on the 6-minute walk distance. J Heart Lung Transplant 2015;34:362–8. http://dx.doi.org/10.1016/j.healon.2014.08.020
37. Gabler NB, French B, Strom BL et al. Validation of 6-minute walk distance as a surrogate end point in pulmonary arterial hypertension trials. Circulation 2012;126:349–56. http://dx.doi.org/10.1161/CIRCULATIONAHA.112.105890
38. Mathai SC, Puhan MA, Lam D, Wise RA. The minimal important difference in the 6-minute walk test for patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 2012;186:428–33. http://dx.doi.org/10.1164/rccm.201203-0480OC
39. Nickel N, Golpon H, Greer M et al. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2012;39:589–96. http://dx.doi.org/10.1183/09031936.00092311
40. Williams MH, Handler CE, Akram R et al. Role of N-terminal brain natriuretic peptide (N-TproBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J 2006;27:1485−94. http://dx.doi.org/10.1093/eurheartj/ehi891
41. van Wolferen SA, Marcus JT, Boonstra A et al. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur Heart J 2007;28:1250−7. http://dx.doi.org/10.1093/eurheartj/ehl477
42. van de Veerdonk MC, Kind T, Marcus JT et al. Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J Am Coll Cardiol 2011;58:2511−19. http://dx.doi.org/10.1016/j.jacc.2011.06.068
43. Dalen JE, Alpert JS. Natural history of pulmonary embolism. Prog Cardiovasc Dis 1975;17:259–70. http://dx.doi.org/10.1016/S0033-0620(75)80017-X
44. Jenkins D, Mayer E, Screaton N, Madani M. Sate-of-the-art chronic thromboembolic pulmonary hypertension diagnosis and management. Eur Respir Rev 2012;21:32–9. http://dx.doi.org/10.1183/09059180.00009211
45. Ribeiro A, Lindmarker P, Johnsson H, Juhlin-Dannfelt A, Jorfeldt L. Pulmonary embolism: one-year follow-up with echocardiograpy Doppler and five-year survival analysis. Circulation 1999;99:1325–30. http://dx.doi.org/10.1161/01.CIR.99.10.1325
46. Becattini C, Agnelli G, Pesavento R et al. Incidence of chronic thromboembolic pulmonary hypertension after a first episode of pulmonary embolism. Chest 2006;130:172–5. http://dx.doi.org/10.1378/chest.130.1.172
47. Miniati M, Monit S, Bottai M et al. Survival and restoration of pulmonary perfusion in a long-term follow-up of patients after acute pulmonary embolism. Medicine (Baltimore) 2006;85:253–62. http://dx.doi.org/10.1097/01.md.0000236952.87590.c8
48. Pengo V, Lensing AW, Prins MH et al. Incidence of chronic throboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004;350:2257–64. http://dx.doi.org/10.1056/NEJMoa032274
49. Marti D, Gomez V, Escobar C et al. Incidence of symptomatic and asymptomatic chronic thromoembolic pulmonary hypertension. Arch Bronconeumol 2010;46;628–33. http://dx.doi.org/10.1016/j.arbres.2010.07.012
50. Klok FA, van Kralingen KW, van Djik AP et al. Prospective cardiopulmonary screening program to detect chronic thromboembolic pulmonary hypertension in patients after acute pulmonary embolism. Haematologica 2010;95:970–5. http://dx.doi.org/10.3324/haematol.2009.018960
51. Bonderman D, Jakowitsch J, Adlbrecht C et al. Medical conditions increasing the risk of chronic thromboembolic pulmonary hypertension. Thromb Haemost 2005;93:512–16. http://dx.doi.org/10.1160/TH04-10-0657
52. Bonderman D, Wilkens H, Wakounig S et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J 2009;33:325–31. http://dx.doi.org/10.1183/09031936.00087608
53. Condliffe R, Kiely DG, Gibbs JS et al. Prognostic and aetiological factors in chronic thromboembolic pulmonary hypertension. Eur Respir J 2009;33:332–8. http://dx.doi.org/10.1183/09031936.00092008
54. Pepke-Zaba J, Delacroix M, Lang I et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 2011;124:1973–81. http://dx.doi.org/10.1161/CIRCULATIONAHA.110.015008
55. Jaïs X, D’Armini AM, Jansa P et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronic Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol 2008;52:2127–34. http://dx.doi.org/10.1016/j.jacc.2008.08.059
56. Simonneau G, D’Armini AM, Ghofrani HA et al. Riociguat for the treatment of chronic throboembolic pulmonary hypertension: a long-term extension study (CHEST-2). Eur Respir J 2014;online first. http://dx.doi.org/10.1183/09031936.00087114
57. Sugiyama M, Fukuda T, Sanda Y, et al. Organized thrombus in pulmonary arteries in patients with chronic thromboembolic pulmonary hypertension; imaging with cone beam computed tomography. Jpn J Radiol 2014;32:375−82. http://dx.doi.org/10.1007/s11604-014-0319-8
