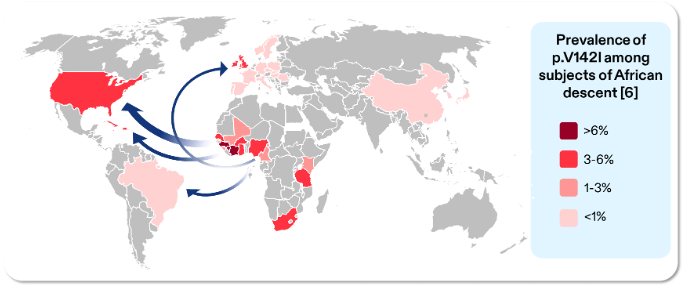
Transthyretin Amyloidosis (ATTR) is a progressive, systemic and life-shortening condition that remains widely under-recognised and frequently misdiagnosed.1 It occurs in two distinct forms: wild-type (wtATTR), which is typically age-related, and hereditary (hATTR), which is caused by pathogenic variants in the transthyretin (TTR) gene.1,2 The p.V142I (formerly V122I) variant of hATTR primarily presents as cardiomyopathy (CM), but can also result in other clinical manifestations such as polyneuropathy.3 The p.V142l variant is most commonly found in individuals with West African and African-Caribbean ancestry.4–7
Individuals with p.V142I typically present with heart failure (HF), restrictive cardiomyopathy, and arrhythmias which can ultimately lead to death.5,11,12
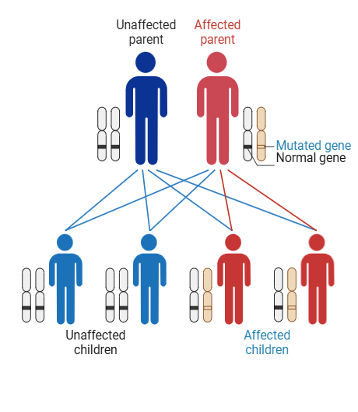
The hereditary form follows an autosomal dominant inheritance pattern. Only one parent needs to pass on the variant for a child to carry it.13–15
Genetic testing is critical – not only to confirm diagnosis and identify family members who carry the variant, but also to enable early detection and timely intervention for improved outcomes.16,17
Individuals with p.V142I typically present with heart failure (HF), restrictive cardiomyopathy, and arrhythmias which can ultimately lead to death.5,11,12
The hereditary form follows an autosomal dominant inheritance pattern. Only one parent needs to pass on the variant for a child to carry it.13–15
Genetic testing is critical – not only to confirm diagnosis and identify family members who carry the variant, but also to enable early detection and timely intervention for improved outcomes.16,17
Figure adapted from: Chandrashekar, P., et al. Prevalence and Outcomes of p.Val142Ile TTR Amyloidosis Cardiomyopathy: A Systematic Review. Circ Genom Precis Med, 2021. 14(5): p. e003356.6
A UK study showed at the time of diagnosis, the p.V124I group of patients has significantly worse LVEF and functional status than the rest of the cohort. Survival was also reduced in the p.V142I group compared to the rest of the subjects.19
This could indicate that the p.V142I variant may present a more aggressive phenotype.19
A US study also showed that black patients, the majority of whom had hATTR-CM caused by p.V142I (102/129, 79%), often presented with more severe disease at onset and poorer survival than their white counterparts who primarily had wild type ATTR-CM (96/135 71.1%).20
A UK study showed at the time of diagnosis, the p.V124I group of patients has significantly worse LVEF and functional status than the rest of the cohort. Survival was also reduced in the p.V142I group compared to the rest of the subjects.19
This could indicate that the p.V142I variant may present a more aggressive phenotype.19
A US study also showed that black patients, the majority of whom had hATTR-CM caused by p.V142I (102/129, 79%), often presented with more severe disease at onset and poorer survival than their white counterparts who primarily had wild type ATTR-CM (96/135 71.1%).20
These delays mean that many patients are only diagnosed when their disease is advanced, with irreversible and extensive cardiac damage,
severely limiting their survival prospects.3,8,22,23 Diagnostic delays are also seen for wild-type patients.19 Another UK study reported that
healthcare costs for ATTR-CM patients are high from symptom onset, rising with disease progression and linked to delayed diagnosis.24
Before diagnosis, none of the participants had heard of hATTR, and described low awareness in primary care. They felt the speed of diagnosis depended heavily on whether they reached a clinician familiar with the condition.
One patient shared how joining a PAG (Amyloidosis UK) provided practical guidance and emotional support, helping them feel “less alone.” They valued hearing from others with lived experience. The rest of the participants, however, were unaware of PAGs. It was suggested that information about PAGs should be included with appointment letters so patients and families can access support early. Healthcare practitioners who implement patient advocacy can achieve greater patient satisfaction and reduce health disparities across all communities they serve.27
Participants described confusion about the genetic testing pathway, including uncertainty about who to contact and how relatives can access testing. While asymptomatic carriers are increasingly identified via genotyping, participants noted the need for clearer guidance on screening and management. Evidence suggests systematic screening can improve early identification and outcomes, reinforcing the need for formalised pathways and guidance.28
The UK Amyloidosis Network should consider providing clear, simple guidance on genetic testing and screening, including referral routes and eligibility.
Disclaimer: Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs become necessary. The editors/authors/contributors and the publishers Medinews (Cardiology) Ltd have taken care to ensure that the information given in this text is accurate and up to date at the time of publication.
Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice. Medinews (Cardiology) Limited advises healthcare professionals to consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.
The opinions, data and statements that appear are those of the contributors. The publishers, editors, and members of the editorial board do not necessarily share the views expressed herein. Although every effort is made to ensure accuracy and avoid mistakes, no liability on the part of the publisher, editors, the editorial board or their agents or employees is accepted for the consequences of any inaccurate or misleading information.
© Medinews (Cardiology) Ltd 2026. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior written permission of the publishers, Medinews (Cardiology) Ltd. It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.