Highlights of the American Heart Association (AHA) Scientific Sessions 2013, held in Dallas, Texas, USA, last November included success with a fourth new oral anticoagulant in patients with atrial fibrillation, and some benefit with spironolactone for heart failure patients with preserved left ventricular function, a group for whom no treatment is currently available.
ENGAGE AF-TIMI 48: success for edoxaban in AF
The new factor Xa inhibitor, edoxaban (Daiichi-Sankyo), was as effective in preventing strokes and safer than warfarin in patients with atrial fibrillation (AF) in the ENGAGE AF-TIMI 48 trial.
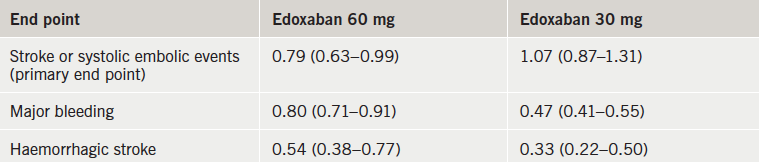
The ENGAGE AF-TIMI 48 (Effective AnticoaGulation with Factor XA Next Generation in Atrial Fibrillation – Thrombolysis In Myocardial Infarction 48) trial included more than 21,000 AF patients from 46 countries who were randomised to edoxaban at one of two doses (60 mg or 30 mg per day) or warfarin.
Results (table 1) showed that both edoxaban doses were associated with significantly less major bleeding than warfarin. The rate of ischaemic stroke was similar with high-dose edoxaban and warfarin but was higher with the low-dose edoxaban regimen. Haemorrhagic strokes and cardiovascular mortality were both significantly lower with both doses of edoxaban than with warfarin.
Dr Robert Giugliano (Brigham and Women’s Hospital, Boston, USA) who was lead investigator of the study, said: “Once-daily edoxaban may be an important alternative to warfarin”.
Designated discussant of the trial, Dr Elaine Hylek (Boston University Medical Center, Boston, USA), highlighted the reduction in haemorrhagic stroke that has been seen with all the new oral anticoagulants in comparison with warfarin. She commented: “The current trial provides very important confirmation for another oral factor Xa inhibitor, that indeed we are seeing a dramatic reduction in intracerebral haemorrhage.”
Compared to previous studies of other new oral anticoagulants, the ENGAGE AF-TIMI 48 trial design stipulated a more rigorous dosage reduction for patients with certain features that enhance blood levels of the drug. This recommended dosage reduction was 50% for patients with renal dysfunction, low body weight, or those who were also taking P-glycoprotein-inhibiting drugs such as verapamil or quinidine. These dosage reductions applied to about a quarter of patients in the trial. “The dosage reduction worked in that it maintained similar efficacy as seen in those patients who did not need to be dose-reduced, and patients with dosage modifications did better on the trial’s measures of safety.” Dr Guigliano said.
Another impressive feature of the trial was that the benefit of edoxaban was compared with fairly well managed warfarin treatment, with warfarin patients being in the therapeutic INR range for 68.4% of the time, Dr Guigliano pointed out.
Edoxaban is now the fourth new oral anticoagulant to have shown benefit in this indication, joining dabigatran, rivaroxaban and apixaban. Answering questions on how clinicians will choose which agent to use, commentators at an AHA media briefing on the ENGAGE AF-TIMI 48 trial said all four new agents seemed to have benefits over warfarin, particularly on safety, but were hard to compare with each other. They suggested that clinicians will make a judgment on patient characteristics that may favour one drug over the others, such as individual side effects of the drugs and dosing schedules.
Edoxaban is currently only available in Japan for patients undergoing orthopedic surgery.
ENGAGE AF-TIMI 48 was also published online in the New England Journal of Medicine on 28th November 2013 (doi: 10.1056/NEJMoa1310907).
STREAM: thrombolysis still an option if no immediate PCI
One-year results from the STREAM trial show similar survival rates between immediate thrombolysis with tenecteplase and transfer to percutaneous coronary intervention (PCI) in ST-elevation myocardial infarction (STEMI) patients for whom PCI is not immediately available.
The 30-day results – showing similar outcomes in the two treatment groups – were presented at the American College of Cardiology (ACC) meeting last March.
STREAM (Strategic Reperfusion Early After Myocardial Infarction) is the first trial to have succeeded in showing that fibrinolysis given before transfer to a PCI hospital can be as effective as primary PCI, which has been attributed to the fact that thrombolysis patients were only given urgent PCI on arrival at the PCI hospital if the electrocardiogram showed they had not reperfused. This avoided the situation of performing PCI with fibrinolysis on board – which has been associated with adverse outcomes – in two-thirds of patients.
The STREAM trial included 1,892 STEMI patients who were not able to undergo PCI within the first hour of arriving at hospital. They were randomised to medical therapy with age-adjusted bolus tenecteplase, clopidogrel and enoxaparin, which was followed by later PCI only if symptoms persisted, or to PCI as soon as it could be performed. Both groups were treated within three hours of symptom onset.
One-year mortality rates were 2.1% in the thrombolysis group versus 1.5% in the PCI group, a non-significant difference.
“In this study, the thrombolytic strategy proved a reasonable approach to take as an initial treatment immediately after severe heart attack when PCI is not immediately available,” said Dr Peter Sinnaeve, the study’s lead author and assistant professor of cardiology (University of Leuven, Belgium).
Using genetics to guide warfarin dose: contradictory results
Two new trials looking at the use of genetic information to guide warfarin dosing have reached different conclusions.
The EU-PACT (European Pharmacogenetics of Anticoagulant Therapy – Warfarin) study, conducted in Sweden and the UK, showed that genetically guided dosing was associated with a greater time in therapeutic range, whereas the COAG (Clarification of Optimal Anticoagulation Through Genetics) trial, conducted in the USA, showed no difference in time in therapeutic range, and a possible worse result for African-American patients, when genetics were used in addition to normal clinical variables.
Trying to address the reason for the different results, lead investigator of the EU-PACT trial, Dr Munir Pirmohamed (University of Liverpool), suggested that this might be due to the specific algorithms used in conjunction with the genetic testing. He pointed out that the algorithm used in EU-PACT was developed and tested in a European population, which was similar to the population included in this EU-PACT study. He said that more work need to be done to develop further ethnicity-specific algorithms to apply to other populations.
In the COAG trial, 1,015 patients with a history of stroke, venous thrombosis or atrial fibrillation (AF), who took warfarin were randomised to dose adjustment using clinical information, such as age, weight, and smoking status, or clinical information plus genetic information on three genes (CYP2C9*2, CYP2C9*3, and VKORC1) that affect warfarin levels. Patients were followed for up to six months from 2009–2013.
Results showed that both groups were within the appropriate therapeutic range 45% of the time over the first four weeks. “These findings highlight the importance of developing and evaluating pharmacogenetic testing in patients from diverse racial and ethnic backgrounds,” said Dr Gary H Gibbons (National Heart, Lung, and Blood Institute, Bethesda, Maryland, USA), which funded the COAG trial.
In the EU-PACT study, 454 patients with either AF or venous thromboembolism were randomised to clinical or genetic tests to guide warfarin therapy. For the genetic group, information about the same three genes as in COAG was used to adjust warfarin doses. At 12 weeks, the genetic guided group had been in therapeutic range for 67% of the time, compared to 60% for those patients guided by clinical factors alone. Dr Pirmohamed estimated the cost of such genetic guidance at about 50 euros per test, and said the next step would be to look at the cost-effectiveness of the process. “My feeling is that we would need to undertake further studies to determine whether this had an impact over the long term in terms of clinical outcome measures,” he commented.
Both studies were published online in The New England Journal of Medicine (EU-PACT doi: 10.1056/NEJMoa1311386; COAG doi: 10.1056/NEJMoa1310669). In an accompanying editorial (doi: 10.1056/NEJMe1313682), Dr Bruce Furie (Harvard Medical School, Boston, USA) concluded that “these trials indicate that this pharmacogenetic testing has either no usefulness in the initial dosing of vitamin K antagonists or, at best, marginal usefulness, given the cost and effort required to perform this testing”.
Repair and replacement of mitral valve give similar results
Patients with severe ischaemic mitral regurgitation (MR) had similar heart function and survival rates whether their mitral valve was repaired or replaced in the CTSN (Cardiothoracic Surgical Trials Network) Severe MR trial. But patientswith repaired valves had a greater risk of experiencing recurrent regurgitation.
“Practice guidelines recommend repairing or replacing mitral valves in severe cases, but there has been a lack of conclusive evidence that one approach is better than the other,” said Dr Michael Acker (University of Pennsylvania’s Perelman School of Medicine, USA) and a clinical investigator in the trial. But he noted that in the USA there is a strong preference for repair over replacement. “But our study suggests that mitral valve replacement provides a more durable correction.”
The study included 251 patients with severe ischaemic mitral valve regurgitation in 22 USA clinical centres. It compared 125 patients who underwent valve replacement with 126 who had their faulty valve repaired.
Results (table 1) showed that rates of death, stroke and re-hopsitalisation for heart failure were similar in the two groups at one year. In addition, there was no significant difference in the mean left-ventricular end-systolic volume index (LVESVI) between groups – 54.6 mL/m2 in the repair group versus 60.7 mL/m2 in the replacement group. This translated into a mean change from baseline of -6.6 and -6.8 mL/m2, respectively.
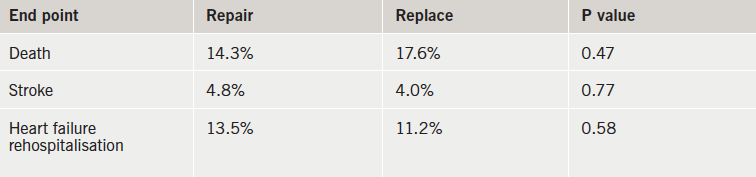
However, the recurrence of mitral regurgitation at 12 months was much higher in the repair group – 32.6% versus 2.3% in the replacement group. In addition, three patients in the repair group underwent reoperation while none of the patients in the replacement group needed a repeat procedure.
TOPCAT: spironolactone shows a signal of benefit
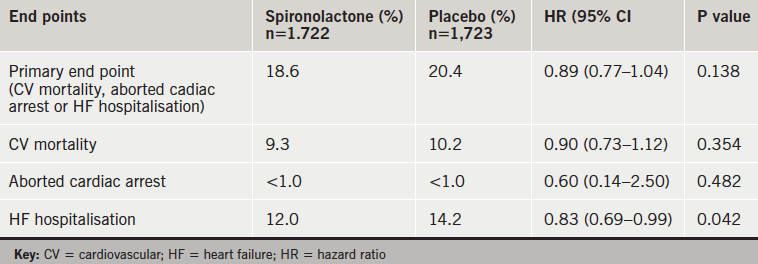
Spironolactone did not show a benefit on the composite primary end point in the TOPCAT trial in patients with heart failure with preserved ejection fraction, but the drug was associated with significantly fewer heart-failure hospitalisations.
Presenting the study, Professor Marc Pfeffer (Harvard Medical School, Brigham and Women’s Hospital, Boston, USA) said the reduction in hospitalisation was “an important finding”.
Noting that at present there is no therapy for patients with heart failure with preserved ejection fraction, Professor Pfeffer said that he would now use spironolactone for this patient population. “We have a generic medication that we can show how to use safely and we do believe it relieves the burden these patients have,” he commented.
Designated discussant Dr Margaret M Redfield (Mayo Clinic, Rochester, USA) agreed that the trial, “although not statistically significant, showed a signal of benefit”. But she also cautioned that the occurrence of worsening renal function and hyperkalaemia would likely be more common in clinical practice given the careful creatinine and potassium monitoring that occurred in the trial.
TOPCAT (Treatment of Preserved Cardiac Function Heart Failure with an Aldosterone Antagonist) included 3,445 heart-failure patients aged 50 or over with an LVEF >45% from six countries. They were randomised to spironolactone (titrated up to 30–45 mg/day) or placebo. The mean follow up was 3.3 years.
There were no significant differences in clinical adverse events (table 1), but there was more hyperkalaemia with spironolactone and more hypokalaemia on placebo (table 2). There were no hyperkalemia-related deaths.

Bleeding risk with new anticoagulants varies with indication
While there appears to be little difference in overall bleeding risks between the various new oral anticoagulants, there does appear to be variations based on in which indication they are being used, a new meta-analysis has found.
The analysis was based on 48 randomised clinical trials including a total of 141,932 patients testing dabigatran, rivaroxaban, apixaban, edoxaban or darexaban in various clinical indications.
Presenting the results, Dr Partha Sardar (New York Medical College, USA) noted that as a group, the new oral anticoagulants caused significantly less major bleeding compared with vitamin K antagonists (relative risk 0.82), with no differences between drug type. But while no differences in bleeding were seen between the newer agents and comparators (vitamin K antagonists or low-molecular-weight heparin) for the treatment of atrial fibrillation and extended treatment of venous thromboembolism, significant bleeding differences were seen in other settings.
The new drugs were linked with significantly higher risks of bleeding than the comparator agents in patients undergoing hip surgery, patients with acute coronary syndrome, and medically ill patients being treated for thromboprophylaxis. There was a lower risk of bleeding in patients treated for venous thromboembolism and pulmonary embolism.
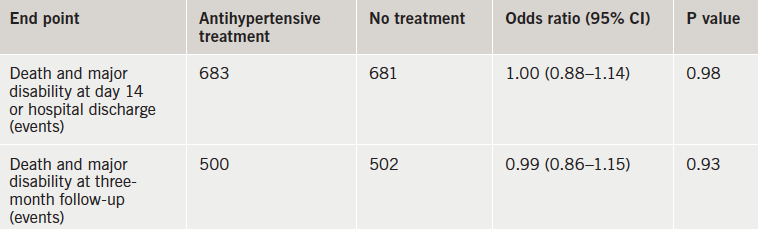
CATIS: no benefit of blood pressure lowering in acute stroke
There was no benefit of antihypertensive treatment in the first days after an acute ischaemic stroke in patients with elevated blood pressure (BP) in the CATIS (China Antihypertensive Trial in Acute Ischaemic Stroke) trial.
“These findings suggest that unless a patient’s systolic BP is 220 mmHg or more or diastolic pressure is 120 mmHg or more, the decision to lower BP with antihypertensive treatment in patients with acute ischaemic stroke does not improve or worsen outcome and therefore should be based on individual clinical judgment,” the researchers conclude.
The study was published online on 17th November 2013 in JAMA (doi: 10.1001/jama.2013.282543) to coincide with its presentation at the AHA meeting.
The authors explain that although the benefit of antihypertensive treatment in reducing the risk for stroke in both primary and secondary prevention is well established, the benefit of reducing BP in the acute phase of stroke is not clear.
The CATIS trial included 4,071 patients from 26 hospitals with acute ischaemic stroke who were within 48 hours of symptom onset and had elevated BP. Patients who received thrombolytic therapy were excluded. Patients were randomised to receive antihypertensive treatment or to discontinue all hypertensive medications they had been on prior to their stroke during their hospitalisation. In those randomised to receive antihypertensive treatment, the aim was to lower BP by 10–25% within 24 hours of randomisation, achieve a BP <140/90 mmHg within seven days, and maintain at this level during hospitalisation. At hospital discharge, all patients were prescribed antihypertensive medications according to clinical guidelines.
Several antihypertensive agents, including intravenous ACE inhibitors (first line), calcium blockers (second line), and diuretics (third line) could be used individually or in combination in the intervention group according to a prespecified treatment algorithm.
While systolic BPs were significantly lowered in the treated group versus the control group by seven days – 137 vs. 146 mmHg – this did not affect outcomes, either on the primary or the secondary composite end point (table 1)