Introduction
The purpose of this module is to review some of the major pathophysiological mechanisms that underlie heart valve disease.1 Whilst these account for the vast majority of patients seen on a clinical basis, it should be remembered that there are numerous rarer causes of valve disease that are beyond the scope of this review. These include congenital defects and acquired conditions related to an array of systemic diseases, tumours, toxins and drugs.
Normal valvular structure
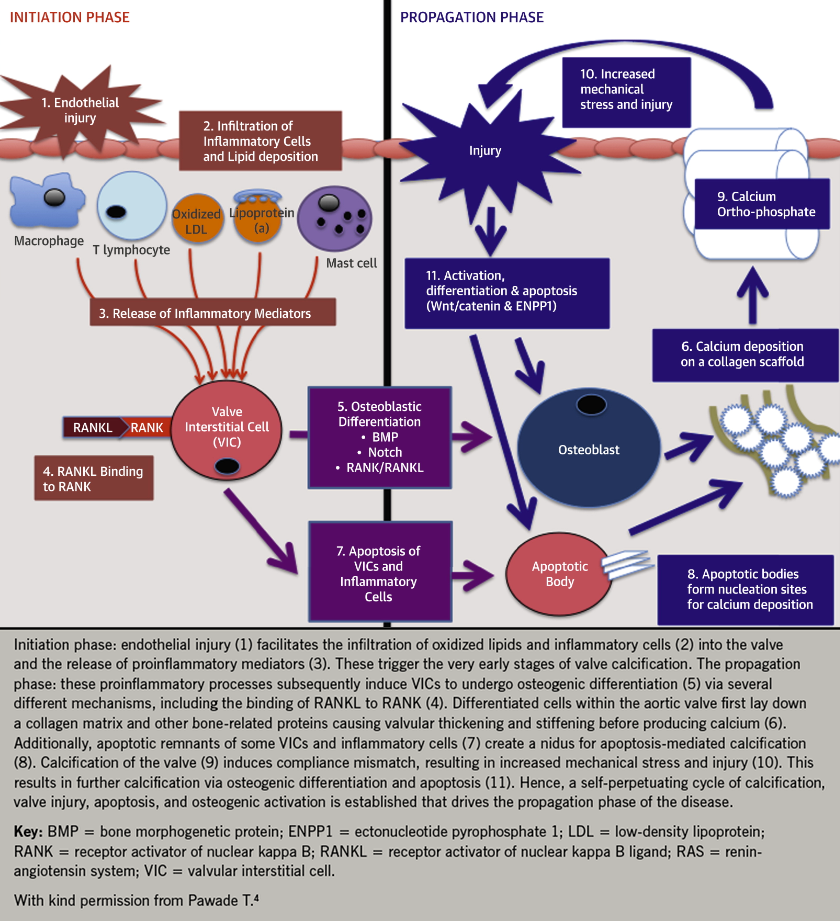
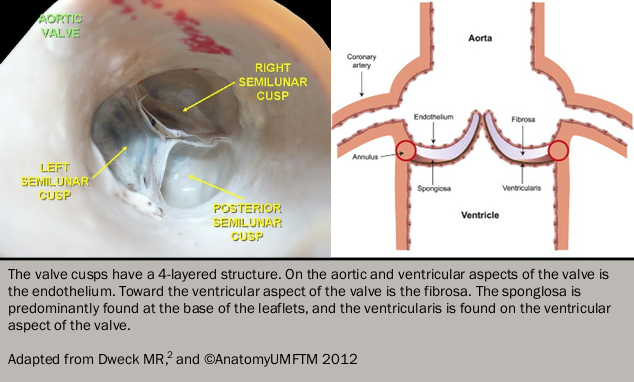
All four cardiac valves share similarities in their basic structure and histological appearances. Externally the leaflets are lined with endothelium and consist of two central layers: the fibrosa and spongiosa. The fibrosa provides strength and structural support for the valve, consisting predominantly of fibroblasts, collagen and elastin fibres. In contrast, the spongiosa is soft and compressible, consisting of mucopolysaccharides and mesenchymal cells, which absorb compressive forces within the valve and providing flexibility to allow the leaflets to change conformation as they open and close (see figure 12). The function of the atrioventricular (mitral and tricuspid) valves also depends on the actions of the chordae tendinae: tendon-like structures consisting of collagen and elastin fibres covered with endothelia which maintain valve competency during ventricular systole.
Calcific valve disease
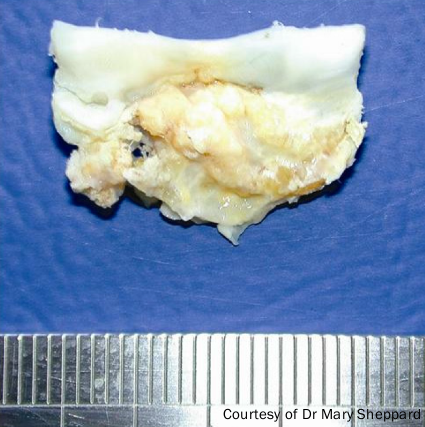
This is perhaps best exemplified by calcific aortic stenosis (see figure 2). Once believed to be the result of prolonged wear and tear and an inevitable consequence of ageing, this common condition is now recognised to be an active, highly-regulated inflammatory condition with common risk factors and similar pathological characteristics to atherosclerosis.3
Aortic stenosis
Progressive narrowing of the aortic valve results from increased thickening and stiffness of the valve leaflets, which restricts their opening and imposes an increased afterload on the left ventricle. Mechanical stress is believed to act as the initiating trigger. This causes damage to the endothelium on the aortic aspect of the valve, facilitating the infiltration of inflammatory cells and lipid. This is believed to be a two stage process, with an initiation phase resulting from endothelial injury and inflammatory cell infiltration and a propagation phase resulting in progressive calcification.4 The pathophysiology is shown in figure 3.4