Systemic inflammatory diseases (SIDs) can present with a wide range of cardiac manifestations, which, although uncommon, are frequently associated with significant morbidity and poor prognosis. Behçet’s disease and antiphospholipid syndrome (APS) are two distinct immune-mediated disorders encompassed within this spectrum, both capable of causing intracardiac thrombi and systemic embolisation, which causes diagnostic and therapeutic challenges. While Behçet’s disease is a classic systemic inflammatory vasculitis, APS primarily represents a prothrombotic autoimmune disorder with variable inflammatory features. This case series highlights two patients with cardiac involvement in SIDs, emphasising the importance of early recognition, individualised treatment strategies, and a multi-disciplinary approach to optimise outcomes in these complex clinical scenarios.
Introduction
Systemic inflammatory diseases (SIDs) are syndromes characterised by constitutional symptoms and multi-organ involvement. Cardiac manifestations are rare but often indicate a poor prognosis.1
Behçet’s disease is a relapsing inflammatory condition presenting with recurrent aphthous stomatitis, genital ulcers, and uveitis. Cardiac involvement occurs in 1–5% of cases and includes myocarditis, conduction abnormalities, and intracardiac thrombi.2
Antiphospholipid syndrome (APS) is an autoimmune disorder associated with thrombotic events and recurrent pregnancy loss. Its cardiac manifestations range from accelerated atherosclerosis and valvular disease to intracardiac thrombi and pulmonary hypertension.3
Case 1
A 41-year-old man with a history of Behçet’s disease presented to the emergency department with a five-day history of fever and constitutional symptoms. Initially admitted for unexplained fever, he was started on antibiotic therapy, but continued to experience febrile episodes. Physical examination revealed no significant abnormalities.
Laboratory tests showed elevated inflammatory markers: C-reactive protein (CRP) of 16.3 mg/dL (normal <0.5 mg/dL) and erythrocyte sedimentation rate (ESR) of 99 mm/hr (normal <15 mm/hr).
Transthoracic echocardiography revealed preserved left ventricular systolic function, but identified an echodense, heterogeneous mass in the right ventricular cavity (figure 1). Cardiac magnetic resonance imaging (MRI) provided further characterisation of this finding, demonstrating a non-enhancing, avascular mass with low signal intensity on both T1- and T2-weighted sequences, and absence of perfusion or late gadolinium enhancement — features consistent with thrombus (figure 1). Chest computed tomography (CT) revealed bilateral pulmonary nodules with a vascular distribution, suggestive of embolic events. Positron emission tomography (PET) showed vascular hypermetabolism in the left lower limb and pulmonary hila, consistent with vasculitis.
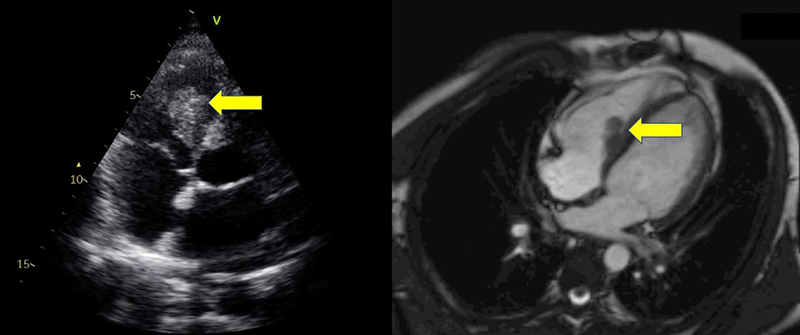
As the patient maintained fever and elevation of systemic inflammatory markers, an exacerbation of his previously diagnosed Behçet’s disease with vascular and cardiac involvement was suspected. This clinical assessment was supported by the international criteria for Behçet’s disease (ICBD).
The patient was treated with unfractionated heparin, corticosteroids (prednisolone 1 mg/kg), and ciclosporin (750 mg/m², up to 1,000 mg daily). Following treatment initiation, the patient’s fever subsided, and inflammatory markers significantly decreased. Echocardiography prior to discharge showed a marked reduction in thrombus size.
Follow-up in a rheumatology clinic included repeat echocardiograms, confirming complete thrombus resolution. The patient experienced new exacerbations of Behçet’s disease, but without recurrence of cardiac manifestations.
Case 2
A 38-year-old woman with a known history of APS, recurrent miscarriages, and prior pulmonary thromboembolism, presented to the emergency department with intermittent retrosternal chest pain. She had been on long-term anticoagulation with warfarin, which had been temporarily replaced by enoxaparin during a recent pregnancy that concluded with delivery, four weeks prior to presentation. At the time of admission, she was on enoxaparin and hydroxychloroquine.
The electrocardiogram (ECG) was unremarkable, and transthoracic echocardiogram revealed normal biventricular function, without regional wall motion abnormalities, with normal valvular structure and function. Laboratory tests showed leukocytosis (19.2 × 10⁹/L, normal 4.5–11.0 × 10⁹/L), elevated CRP (25.3 mg/dL, normal <0.5 mg/dL), and high-sensitivity troponin I of 2,081 pg/ml (normal <47.3 pg/ml). A urinalysis revealed proteinuria in the nephrotic range, with total protein levels of 4,253 mg/L.
Thoracoabdominal CT excluded pulmonary embolism, but revealed findings suggestive of hepatic infarction, and pancreatic calcifications consistent with chronic pancreatitis (figure 2B). Coronary angiography was subsequently performed and showed no obstructive coronary artery disease. Given the suspicion of embolic phenomena, transoesophageal echocardiography was performed to further evaluate for intracardiac sources of embolism. It identified a mass in the superior vena cava extending into the right atrium (figure 2A). Cardiac MRI demonstrated late gadolinium enhancement (subendocardial) in the basal segments of the inferolateral and inferoseptal walls (figure 2C), consistent with an embolic event.
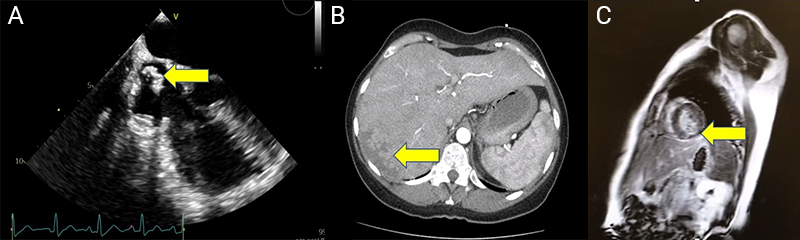
Multi-disciplinary management involved rheumatology and nephrology consultations. The patient received unfractionated heparin initially, which was later transitioned back to warfarin. She maintained treatment with hydroxychloroquine. Renal biopsy was not performed because the patient progressed with normalisation of urinary protein levels. The patient’s symptoms resolved without recurrence of chest pain or disease exacerbations. She remained on warfarin and was monitored in rheumatology and cardiology follow-up.
Discussion
Major cardiac involvement in SIDs is uncommon, but often associated with poor prognosis. Due to its rarity, clinicians may be unfamiliar with the full spectrum of cardiac manifestations in these conditions. Cardiac involvement in SIDs can present as pericarditis, myocarditis, myocardial fibrosis, myocardial ischaemia, heart valve disease, intracardiac thrombi, conduction system disease, pulmonary hypertension, or arterial hypertension.
Intracardiac thrombus is a relatively frequent finding in clinical practice, requiring differentiation from other intracardiac masses or vegetations. Anticoagulation remains the cornerstone of therapy for thrombotic lesions in most cases. However, adjunctive therapies tailored to the underlying condition are often critical. In APS, superior vena cava thrombi, although rare, have been reported.
The choice of anticoagulant should be guided by the underlying systemic condition. In APS, long-term anticoagulation with vitamin K antagonists (e.g. warfarin) is recommended, particularly in high-risk patients with triple positivity — the simultaneous presence of lupus anticoagulant, anti-cardiolipin, and anti-beta2 glycoprotein I antibodies — or a history of thrombotic events. Direct oral anticoagulants (DOACs) are generally not advised in this setting due to higher recurrence rates of thrombosis, as demonstrated in recent trials.4,5
In contrast, in Behçet’s disease, thrombosis is thought to result primarily from vascular inflammation, rather than a primary hypercoagulable state. While anticoagulation is often considered an important component of management, its use remains controversial and should be individualised. Immunosuppressive therapy is regarded as the first-line treatment, aiming to control the underlying inflammation. Anticoagulation is typically reserved for selected cases with extensive or recurrent thrombosis, and must be balanced carefully against bleeding risk, particularly in the presence of pulmonary artery aneurysms.6
Conclusion
This case series highlights the importance of recognising cardiac involvement in SIDs, which, though infrequent, can lead to significant morbidity. Early diagnosis and a multi-disciplinary approach are key to improving outcomes in these patients.
Key messages
Multi-modal imaging for cardiac masses in systemic inflammatory diseases (SIDs). Cardiac involvement in SIDs can manifest as intracardiac thrombi, which may be mistaken for tumours or infective endocarditis. This case series highlights the importance of integrating echocardiography, cardiac magnetic resonance imaging (MRI), and positron-emission tomography (PET) to differentiate thrombotic lesions from other cardiac masses, guiding appropriate treatment strategies
Systemic embolisation in antiphospholipid syndrome (APS) beyond common sites. The second case illustrates that APS-related embolic events can extend beyond the classical pulmonary and cerebral territories, affecting organs such as the liver and pancreas. Recognising these atypical presentations is critical for timely diagnosis and intervention
The role of immunosuppression in Behçet’s disease with cardiac involvement. While anticoagulation is the core treatment for intracardiac thrombi, this case series emphasises that in Behçet’s disease, immunosuppressive therapy (corticosteroids and ciclosporin) plays a crucial role in thrombus resolution and preventing recurrence. This highlights the need for a multi-disciplinary approach, integrating rheumatology and cardiology expertise in management decisions
Conflicts of interest
None declared.
Funding
None.
Patient consent
Patient consents were obtained for the use of images, writing of the case and publication.
References
1. Knockaert DC. Cardiac involvement in systemic inflammatory diseases. Eur Heart J 2007;28:1797–804. https://doi.org/10.1093/eurheartj/ehm193
2. Sakane T, Takeno M, Suzuki N, Inaba G. Behçet’s disease. N Engl J Med 1999;341:1284–91. https://doi.org/10.1056/NEJM199910213411707
3. Soltész P, Szekanecz Z, Kiss E, Shoenfeld Y. Cardiac manifestations in antiphospholipid syndrome. Autoimmun Rev 2007;6:379–86. https://doi.org/10.1016/j.autrev.2007.01.003
4. Devreese KMJ, Bertolaccini ML, Branch DW et al. An update on laboratory detection and interpretation of antiphospholipid antibodies for diagnosis of antiphospholipid syndrome: guidance from the ISTH-SSC Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibodies: reply. J Thromb Haemost 2025;23:1732–4. https://doi.org/10.1016/j.jtha.2025.02.018
5. Pengo V, Denas G, Zoppellaro G et al. Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018;132:1365–71. https://doi.org/10.1182/blood-2018-04-848333
6. Hatemi G, Christensen R, Bang D et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis 2018;77:808–18. https://doi.org/10.1136/annrheumdis-2018-213225
