Highlights of the American College of Cardiology (ACC) meeting held in San Francisco last month included the first study to show a benefit of treating ST-elevation myocardial infarction (STEMI) patients in community hospitals with fibrinolysis before transfer for percutaneous coronary intervention (PCI), as well as evidence supporting the use of PCI in hospitals without surgical back up. There were also positive results for eplerenone in MI, ranolazine in diabetes, and a new IV anti-platelet agent – cangrelor – in PCI. But perhaps the biggest story of the meeting was the removal of the PREVAIL trial from the programme at the last minute because of an embargo break by the sponsor.
PREVAIL not presented but eases safety concerns on Watchman
 The PREVAIL trial of a new device which closes the left atrial appendage in the heart (Watchman®, Boston Scientific) attracted huge controversy at the ACC meeting when it was removed from the programme within an hour of its presentation because of an embargo break by the sponsor, Boston Scientific.
The PREVAIL trial of a new device which closes the left atrial appendage in the heart (Watchman®, Boston Scientific) attracted huge controversy at the ACC meeting when it was removed from the programme within an hour of its presentation because of an embargo break by the sponsor, Boston Scientific.
But the slides and a press release were still made available to the media, and preliminary results appear to suggest some reassurance on safety concerns generated in a previous study.
The device, which is implanted via a trans-septal catheter-based delivery system, is already available in Europe for the prevention of stroke in atrial fibrillation (AF) patients, but it has not been approved by the USA Food and Drug Administration (FDA) because of safety concerns raised in the previous PROTECT-AF trial. These related to a high initial rate of pericardial effusions and procedure-related strokes. The PREVAIL trial was therefore conducted to give more information on safety and to confirm the efficacy results shown in PROTECT-AF.
The PREVAIL trial enrolled 407 patients who were randomised 2:1 to the device or warfarin. Device patients were given 45 days of warfarin therapy. The study had a Bayesian design, which means that the PROTECT results were also taken into account when assessing efficacy. Results showed that the device had a 95.1% implant success rate, up from the 91% rate in PROTECT.
The trial had three co-primary end points, one for safety and two for efficacy. The safety end point was met, as was one of the efficacy end points; the other efficacy end point was narrowly missed. The efficacy results are, however, very preliminary, with only 58 device patients and 30 controls having reached the 18 month follow-up time.
The main safety end point – acute (seven-day) occurrence of death, ischaemic stroke, systemic embolism and procedure, or device-related complications requiring major cardiovascular or endovascular intervention – occurred in six out of 269 patients (2.2%) who received the device. A second, broader, safety end point, including cardiac perforation, pericardial effusion with tamponade, ischaemic stroke, device embolisation, and other vascular complications, occurred in 4.4% of patients receiving the device in PREVAIL, compared with 8.7% in PROTECT.
Cardiac perforation requiring surgical repair occurred in 0.4% of PREVAIL patients receiving the device compared with 1.6% in PROTECT-AF. Pericardial effusion with cardiac tamponade requiring pericardiocentesis occurred in 1.5% of Watchman patients in PREVAIL vs. 2.4% of those in PROTECT AF.
Speaking to the media, lead investigator Dr David Holmes (Mayo Clinic, Minnesota, USA) said: “Despite inclusion of higher-risk patients in PREVAIL than in PROTECT, there were fewer complications, and results show the device can be safely implanted by new operators. The Watchman device therefore offers an alternative to oral anticoagulation therapy for thromboembolic prevention in patients with nonvalvular AF”.
Independent commentator, Dr Gordon Tomaselli (Johns Hopkins University, Baltimore, USA), said he was reassured by the safety data in PREVAIL, and said he would use the device in patients at high risk of stroke who can’t take anticoagulant drugs because of bleeding issues. He added that there were not enough data yet to recommend its use in patients who could take anticoagulants.
HPS-THRIVE shows no reduction in events with niacin
Full results of the large-scale HPS2-THRIVE (Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events) trial have shown no success in the use of extended-release niacin and the antiflushing agent laropiprant for the reduction of major vascular events in patients with well-controlled low-density lipoprotein (LDL) levels, but an increased rate of adverse effects.
The primary end point of the 25,000 patient trial, the combination of coronary death, nonfatal MI, stroke, or coronary revascularisation occurred in 15.0% of patients in the control arm and 14.5% of patients in the niacin/laropiprant arm, a difference that was not statistically significant. There was a 10% reduction in the risk of coronary revascularisation with niacin and laropiprant that just reached statistical significance, but none of the other components of the primary end point reached significance.
In terms of adverse events, the niacin/laropiprant combination was associated with a significant 3.7% absolute excess risk of diabetic complications, a significant 1.8% excess risk of new-onset diabetes, as well as an excess 1.4% higher risk of infection and a 0.7% higher risk of bleeding, including an increased risk of haemorrhagic stroke.
Lead investigator Dr Jane Armitage (Oxford University) said: “In light of these findings the role of extended-release niacin for the prevention of cardiovascular disease should be reconsidered.”
Chair of the study, Dr Rory Collins (Oxford University) added that given the side effects of niacin and the increased risk of serious adverse events, the evidence clearly suggests no benefit of niacin therapy. He added that doctors should instead be using higher doses of statins in order to achieve low levels of LDL cholesterol.
Niacin products “under review”
In response to the HPS-THRIVE results, the European Medicines Agency (EMA) has suspended all niacin/laropiprant products from sale. It has also announced a review of nicotinic acid and related substances, saying that the HPS2-THRIVE data may “have implications for medicines containing only nicotinic acid or related substances,” and it plans to issue an opinion on whether the marketing authorisations for these medicines should be maintained, varied, suspended, or withdrawn across the European Union.
Intensive glycaemic control ups heart failure risk
Both intensive glycaemic control and poor glycaemic control were associated with an increased risk of heart failure in diabetes patients in a new case-control study.
The study was conducted in the diabetic population of the GoDARTS registry. Cases were patients with heart failure defined as either a hospital discharge code for chronic heart failure or systolic dysfunction requiring a loop diuretic. They were matched with controls for gender and age at diabetes diagnosis. Cox-regression analysis was used to examine the link between mean HbA1c and time to chronic heart failure, accounting for all possible confounders.
Of 8,890 patients with diabetes, 759 developed heart failure during the study. Those with poor glycaemic control (HbA1c >6.9%) were twice as likely to develop heart failure (odds ratio 2.26; p<0.01).
However, intensive glycaemic control (HbA1c <6%) also appeared to be associated with a similar increased risk for heart failure (OR 2.48; p<0.01).
The incidence of coronary artery disease and myocardial infarction also increased when mean HbA1c was outside the range of 6–7%.
Presenting the results, Dr Helen Parry (University of Dundee) recommended that type 2 diabetes patients should aim for tight glycaemic control, keeping an HbA1c in the range of 6.0% to 6.9% if they want to reduce their risk of heart failure. But she added that achieving this level of control is not easy.
Dr Parry said the mechanism behind heart failure risk with intensive glycaemic control was probably multifactorial. She noted that heart failure has been associated with several classes of diabetes drugs, which are probably used more in this with intensive control, and that patients with low levels of HbA1c more often experience acute hypoglycaemic events.
TACT: no effect of high dose vitamins on CV events
Taking high-doses of oral vitamins did not reduce the risk of cardiovascular events, new results from TACT (Trial to Assess Chelation Therapy) have shown.
The trial, in 1,700 post-myocardial infarction patients, had a two by two factorial design, and has already reported positive results with chelation therapy. For the vitamin investigation, patients were randomised to a treatment including high doses of vitamins A, B, C, D, E, K, niacin, folate, calcium, magnesium, and zinc, or placebo.
Results showed that the vitamin therapy did not significantly reduce the time to first occurrence of death, MI, stroke, coronary revascularisation, or hospitalisation for angina, although there was 11% non-significant reduction.
When the use of vitamin therapy was combined with chelation and compared with patients who received only placebo, there was a statistically significant 26% relative reduction in the risk of the primary end point.
Chelation results published
The chelation part of TACT, which showed an 18% reduction in events for chelation alone versus placebo, was first reported at last year’s American Heart Association meeting. It has now also been published in the March 26 issue of JAMA (http://dx.doi.org/10.1001/jama.2013.2107). However, the study has been dogged with controversy.
In an editorial accompanying the publication (http://dx.doi.org/10.1001/jama.2013.2778), Dr Steven Nissen (Cleveland Clinic, USA) concludes: “Given the numerous concerns with this expensive, federally funded clinical trial, including missing data, potential investigator or patient unmasking, use of subjective end points, and intentional unblinding of the sponsor, the results cannot be accepted as reliable and do not demonstrate a benefit of chelation therapy.
At the ACC meeting, lead researcher, Dr Gervasio Lamas (Mount Sinai Medical Centre, Miami, US) was cautious about how the chelation results should be interpreted. “We’ve moved something from alternative medicine into scientific inquiry and found some results that merit further research. We don’t think the results of any single trial are enough to carry this novel hypothesis into daily use for patients who have had an acute myocardial infarction.”
REMINDER: eplerenone beneficial in MI?
The aldosterone blocker, eplerenone, started within the first day after an acute myocardial infarction (MI) in patients without heart failure or left ventricular dysfunction can improve outcomes, according to the results of the REMINDER trial.
But the trial generated some controversy, in that the only component of the complex composite primary endpoint to be significantly reduced was the level of natriuretic peptides.
The primary endpoint of the study included cardiovascular mortality, rehospitalisation, prolonged heart failure hospitalisation, sustained ventricular tachycardia or ventricular fibrillation, left ventricular ejection fraction (LVEF) <40% after one month, or levels of natriuretic peptides.
REMINDER (Impact of Eplerenone on Cardiovascular Outcomes in Patients Post Myocardial Infarction) included 1,012 STEMI patients, who received eplerenone 25–50 mg/day or placebo. Treatment started within the first 24 hours of symptom onset and preferably before myocardial reperfusion.
At a mean of 10.5 months follow-up, the composite primary end point occurred in 29.6% of the placebo group versus 18.4% in the eplerenone group (p<0.0001). Most of the benefit was attributable to the natriuretic peptide biomarker, which was raised in 25.9% of the placebo groups versus 16% of the eplerenone group (p<0.0002). There were, however, trends in favour of eplerenone for heart-failure rehospitalisation and ventricular arrhythmias.
Safety was good, with similar rates of hyperkalemia in the eplerenone and placebo groups, and hypokalaemia occurring more often in the control group, noted lead investigator Dr Gilles Montalescot (Pitié-Salpêtrière University Hospital, France).
Discussing the trial, Dr Magnus Ohman (Duke University, Kansas City, USA) noted that eplerenone was not well used for heart failure because of concerns about hyperkalaemia. REMINDER could be considered more of a safety study for eplerenone rather than an efficacy study, showing that eplerenone is safe when you exclude patients with renal failure, he said.
Dr Ohman added that the clinical significance of a natriuretic-peptide elevation after 30 days was not clear but that it might be a predictor of clinical outcomes in the future.
MASS COMM: elective PCI safe without surgical back up
Non-emergency percutaneous coronary intervention (PCI) can be performed safely in hospitals without on-site cardiac surgery, according to the results of the MASS COMM (PCI Outcomes in Community Versus Tertiary Settings) study.
The study, which was published online in the New England Journal of Medicine (http://dx.doi.org/10.1056/NEJMoa1300610) to coincide with its presentation at the ACC, included 3,691 patients who presented for elective PCI at a hospital without on-site cardiac surgery capabilities. They were randomised to receive PCI at the community hospital (2,774 patients) or to be transferred to a partner hospital with on-site cardiac surgery backup (917 patients).
Results showed that there was no difference in major adverse cardiac events (death, MI, repeat revascularisation, or stroke) at 30 days and 12 months when these patients were compared with those treated at hospitals with surgical back-up.
Presenting the data, Dr Alice Jacobs (Boston University School of Medicine, Boston, USA) said the results were in line with those of the C-PORT-E (Cardiovascular Patient Outcomes Research Team Elective Angioplasty) study. “These data suggest that PCI can be safely performed at hospitals without surgery on site, albeit with formal programmes and volume requirements, and that this could be an acceptable option for patients presenting to such hospitals,” she noted.
To participate in the study, hospitals had to perform a minimum of 300 diagnostic procedures in each of the previous two years, including 36 primary PCI procedures annually. The hospitals were also required to be within 60 minutes of a centre with 24 hour surgical backup, and physicians had to have performed a minimum of 75 PCI procedures annually.
Rivaroxaban: low risk of short treatment interruptions

Atrial fibrillation patients who stop taking rivaroxaban for a few days had a low risk of strokes or thrombotic events similar to that seen with similar interruptions of warfarin treatment, new results (see table 1) from the ROCKET-AF trial suggest.
In the analysis, a temporary interruption was defined as three days or more. Of the 14,236 patients in the study, 2,165 receiving rivaroxaban and 2,528 of those on warfarin stopped treatment at some point during the trial for an average duration of five days. The at -risk period was defined as three days after the drug was stopped until three days after the drug was restarted.
STREAM: success for fibrinolysis before transport for PCI
Fibrinolysis with tenecteplase and contemporary antithrombotic therapy given before transport to a hospital capable of percutaneous coronary intervention (PCI) is as effective as primary PCI in ST-elevation myocardial infarction (STEMI) patients presenting within three hours of symptom onset, according to the results of the STREAM (Strategic Reperfusion Early After Myocardial Infarction) trial.
Lead investigator, Dr Frans Van de Werf (University of Leuven, Belgium) said: “Fibrinolysis before transfer is at least as effective as PCI, and an urgent catheterisation was avoided in two-thirds of patients. This gives clinicians time to consider other options, such as coronary artery bypass graft (CABG) surgery and medical therapy.”
STREAM differs from other trials investigating fibrinolysis before transfer for PCI in that patients were only given urgent PCI on arrival at the tertiary hospital if they had not reperfused (as judged by the ECG). Because of this, the majority of patients avoided having to undergo PCI with fibrinolysis on board which has been associated with adverse outcomes in pervious studies.
Discussing the study, which was published online in the New England Journal of Medicine (http://dx.doi.org/10.1056/NEJMoa1301092) to coincide with the presentation, Dr Freek Verheugt (Nijmegen University, the Netherlands) said: ” I think this will change practice in certain parts of the world where there are long driving times to PCI hospitals.”
The trial involved 1,915 STEMI patients from 15 countries who were first seen in community hospitals or by emergency medical personnel, and for whom there was going to be a delay of over one hour before PCI could be performed. Before transfer, patients were randomised to PCI immediately after arrival or to tenecteplase plus enoxaparin, clopidogrel, and aspirin before arrival. Elderly patients had lower doses of clopidogrel (no loading dose) and enoxaparin.
When patients on tenecteplase reached a major medical centre, 36% needed urgent PCI. The other 64% of patients did not need urgent PCI and received an angiogram an average of 17 hours after arrival and, based on the results, received PCI or CABG under non-urgent circumstances.

The primary end point (see table 1) – a composite of all-cause mortality, shock, congestive heart failure, and subsequent heart attack at 30 days – were similar between the immediate-PCI and tenecteplase groups.

There was a significant increase in intracranial haemorrhage (ICH) in the fibrinolysis group, which led to the dose of tenecteplase being halved in people aged 75 years and older fairly early on in the course of the trial, after which the intracranial haemorrhage rate in the fibrinolysis group was reduced to 0.5% – not significantly different from the PCI group (see table 2). After the dosage amendment was made in the elderly, the primary end point almost reached significance in favor of the fibrinolysis group, Dr Van de Werf noted.
TERISA: ranolazine reduces angina in diabetics
The anti-anginal drug, ranolazine, is effective at reducing angina episodes specifically in patients with diabetes, the TERISA (Type 2 Diabetes Evaluation of Ranolazine in Subjects With Chronic Stable Angina) trial has shown.
Lead investigator, Dr Mikhail Kosiborod (Saint Luke’s Mid America Heart Institute, Kansas City, USA) noted that ranolazine is already approved as an anti-anginal but it was important to demonstrate a benefit in the particularly challenging diabetes population.
The study, which was simultaneously published online in the Journal of the American College Cardiology (http://dx.doi.org/10.1016/j.jacc.2013.02.011) to coincide with its ACC presentation, involved 949 type 2 diabetes patients who were randomised to a target dose of ranolazine 1,000 mg twice a day or placebo, for eight weeks. These patients had a high burden of angina, with around six to seven episodes per week, and were already receiving treatment with one or two anti-anginals.
The primary endpoint – average weekly angina frequency between weeks 2 and 8 – was significantly improved in the ranolazine group (3.8 episodes vs. 4.3 per week; p=0.008). Weekly use of rescue sublingual nitroglycerin was also lower among the ranolazine-treated patients (1.7 vs. 2.1 doses per week; p=0.003).
An editorial accompanying the publication (http://dx.doi.org/10.1016/j.jacc.2013.03.002) notes that the results confirm prior post hoc analyses of subgroups from the CARISA (Combination Assessment of Ranolazine in Stable Angina) and MERLIN-TIMI 36 (Metabolic Efficiency With Ranolazine for Less Ischemia in Non-ST-Elevation Acute Coronary Syndromes) studies. But it adds that the absolute effects, although statistically significant, were small, with only 0.5 fewer episodes of angina and 0.4 fewer sublingual nitroglycerin tablets used per week. “The clinical relevance of such slight absolute differences may be questioned,” it concludes.
CHAMPION-PHOENIX: cangrelor looks good for acute use in PCI
The new intravenous antiplatelet agent, cangrelor, reduced the risk of periprocedural complications of percutaneous coronary intervention (PCI) compared with clopidogrel in CHAMPION-PHOENIX (Clinical Trial Comparing Cangrelor to Clopidogrel Standard Therapy in Subjects Who Require PCI).
The trial, which was simultaneously published online in the New England Journal of Medicine (http://dx.doi.org/10.1056/NEJMoa1300815), randomised 11,145 patients undergoing urgent or elective PCI to receive cangrelor (30 µg/kg bolus followed by a 4 µg/kg/min infusion for at least two hours) or a 600-mg or 300-mg loading dose of clopidogrel.
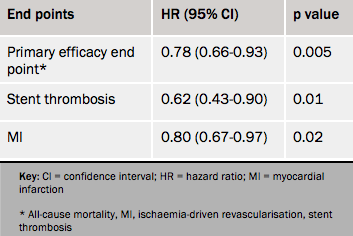
Results (see table 1) showed a 22% reduction in the composite efficacy end point with cangrelor, driven by reductions in myocardial infarction and stent thrombosis.
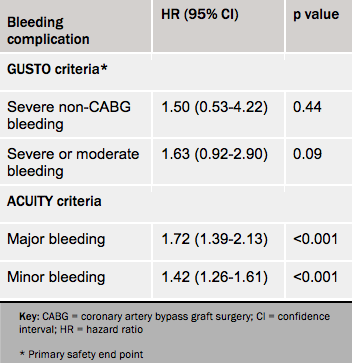
Bleeding complications as prospectively defined according to the GUSTO criteria were not increased, but when defined using the more sensitive ACUITY definitions, there was more bleeding with cangrelor than with clopidogrel (see table 2).
Co-principal investigator, Dr Deepak L Bhatt (Brigham and Women’s Hospital, Boston, USA) told a press conference that one of the main advantages of cangrelor was its short-acting nature which gave the flexibility to initiate and rapidly stop platelet inhibition, in case the patient requires surgery or develops bleeding complications.
However, an editorial accompanying the publication (http://dx.doi.org/10.1056/
NEJMe1302504) points out some problems with the study – namely that the full antiplatelet effects of cangrelor were operative before and during PCI, whereas 37% of patients in the clopidogrel group received the drug during or after PCI; and that many patients received clopidogrel at the inferior 300 mg dose. Given these points, the routine use of cangrelor for all patients undergoing PCI is not yet justified, it concludes.
