Syndromes and pathophysiology
Over many years, the heart failure syndrome has attracted a number of different terminologies of varying degrees of usefulness. It makes sense, perhaps, to consider the separate heart failure syndromes as the pathophysiology of them is distinct: the differences highlight important differences in therapeutic approach.
The most useful distinction to make is between acute and chronic heart failure. However, even then, the terms can be confusing. Here, we will take acute heart failure to describe patients with sufficiently severe symptoms and signs that they present acutely seeking medical attention (and are often admitted to hospital); whereas chronic heart failure describes the syndrome patients have once acute heart failure has been medically treated.
Acute heart failure
The majority, perhaps as many as two thirds, of patients with heart failure are initially diagnosed during a hospitalisation. Acute heart failure is a problem of fluid distribution. Broadly, patients divide into those with acute pulmonary oedema, where there is fluid in the airspaces in the lung, and those with peripheral oedema, where the problem is fluid retention.
Pulmonary oedema: Pulmonary oedema is an acute medical emergency. Patients present with severe breathlessness which is usually of abrupt onset. The experience for the patient is terrifying: she has to sit upright, is barely able to speak and often fears that she will die.
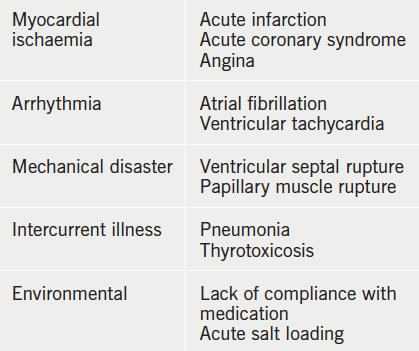
The breathlessness is often accompanied by wheeze and cough productive of pink frothy sputum. There is usually an acute precipitant that has triggered the episode of illness (table 4).
On examination there is massive sympathetic nervous system activation, and the patient is thus usually pale and clammy, with a tachycardia and often hypertension. There is usually a gallop rhythm and widespread crackles and wheezes through the lung fields.
Pathophysiology: Pulmonary oedema is best understood in haemodynamic terms. Fluid is held in the pulmonary capillaries by the balance between the hydrostatic pressure in the capillary tending to push fluid out and the colloid osmotic pressure (largely generated by plasma proteins) tending to hold fluid in the vascular space. There is a net (small) constant transudation of fluid from the pulmonary capillaries which is removed by the lymphatics.
Left ventricular work (and cardiac output) is determined by left ventricular filling pressure (that is, end-diastolic pressure). This is the Frank-Starling relation (figures 9 and 10): as filling pressure rises, so does cardiac output. In the acutely failing left ventricle, the relation is shifted downward and to the left so that to maintain any given cardiac output, a higher filling pressure is required.
As the filling pressure is the same as the pulmonary venous pressure, the rise results in an increase in the rate of transudation from pulmonary capillaries into the lung tissues, and the rate eventually exceeds the rate at which fluid can be removed by the lymphatics. At this point, fluid accumulates in the interstitium of the lung and then the alveoli.
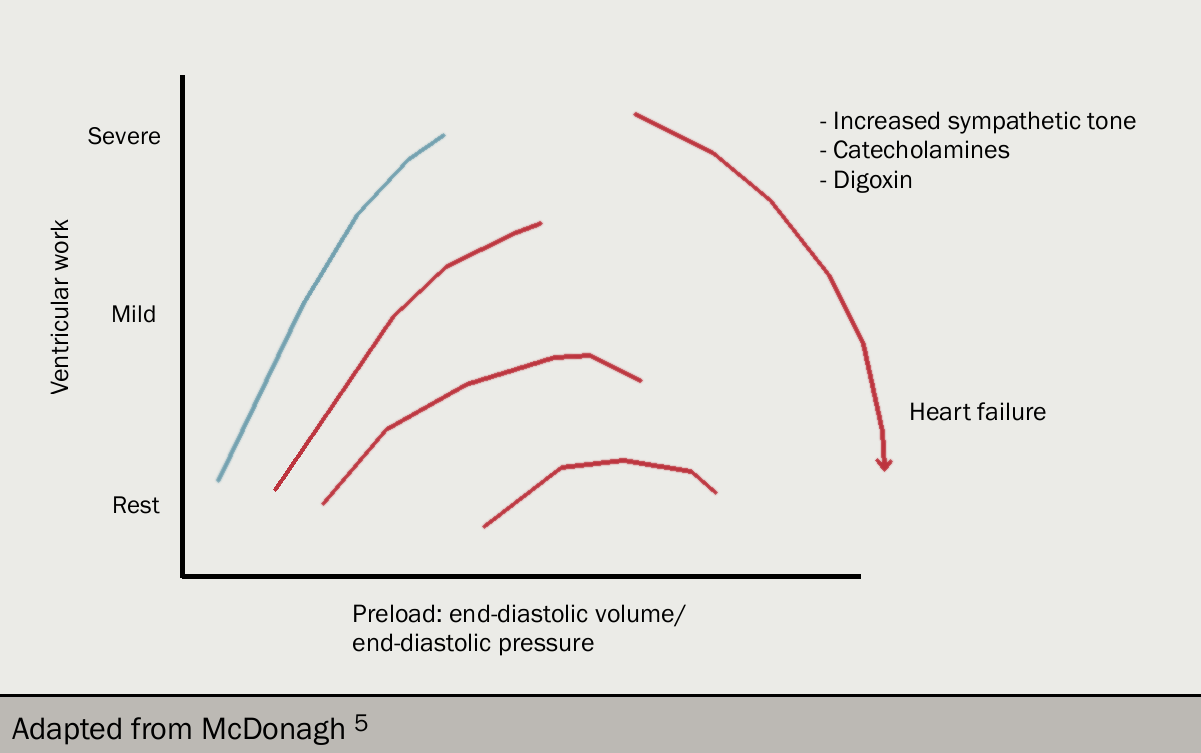

A point often ignored is that the increase in filling pressure requires an input of energy: this can only come from the right ventricle. In acute pulmonary oedema, there must be a temporary imbalance between the output of the two ventricles: the relative excess output from the right results in the increase in left ventricular diastolic pressure and represents the fluid accumulating in the lungs.
Peripheral oedema: More common than pulmonary oedema as a cause of hospitalisation is fluid retention (table 5). In contrast with patients with pulmonary oedema (who may have a low circulating volume due to accumulation of fluid in the lungs), patients with peripheral oedema have an absolute excess of body fluid and an increased circulating volume. The fluid tends to accumulate gradually over many days or weeks; it takes around 5L of excess fluid before peripheral oedema appears.
The order of deposition of oedema is dictated by gravity, so for most patients it starts in the ankles and works upward – and may involve the abdominal, and even thoracic, wall. Patients are usually not breathless at rest, although are unable to do much exercise.
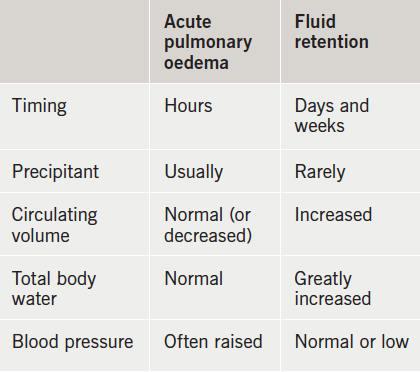
On examination the patient may have a tachycardia with a low volume pulse and a low blood pressure. Around 25% will be in atrial fibrillation. The jugular venous pressure is invariably raised, but may be so high that the top of the column of blood cannot be seen even with the patient sitting upright. Pitting oedema usually starts in the feet and ankles, but be wary of the bed-bound patient in whom sacral oedema may be prominent. In most patients, the heart is dilated and there is a loud third and often fourth heart sound. Some degree of pulmonary venous hypertension is almost invariable, and patients usually have basal crackles in both lung fields.
Pathophysiology: Peripheral oedema develops in much the same was as pulmonary oedema. The increased circulating volume causes in an increase in hydrostatic pressure (with gravity ensuring that the hydrostatic pressure is highest in the feet). The capacity of the lymphatics to drain away the transudate is exceeded, and oedema fluid starts to form.
What is less certain is why there is salt and water retention in the first place. It may be related to the primary need for the body to maintain blood pressure: if, as a consequence of the failing heart, blood pressure falls, the resultant neurohormonal activation (involving renin-angiotensin-aldosterone system [RAAS], antidiuretic hormone and sympathetic system activation) causes salt and water retention in the kidneys. Additionally, reduced renal perfusion is another stimulus to renin production.
This cannot be the whole explanation as in some patients oedema develops despite normal blood pressure; and in others, there is fluid retention despite aggressive treatment with neurohormonal antagonists.

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.