Introduction
Table 1. Common electrolyte abnormalities in HF and their prevalence
| Electrolyte abnormality | Prevalence | N |
| Hyponatraemia <135 mmol/L |
Acute HF: 19.5%10 Chronic HF: 8.2%8 |
4,449 7,173 |
| Hypokalaemia <3.5 mmol/L |
Acute HF: 4.9%4 Chronic HF: 1.7%5 |
2,596 6,073 |
| Hyperkalaemia >5.5 mmol/L |
Acute HF: 4.4%4 Chronic HF: 2.6%4 |
4,449 7,173 |
| Hypochloraemia <96 mmol/L |
Acute HF: 13%6 Chronic HF: 13%7 |
2,033 2,699 |
| Key: HF = heart failure | ||
As well as dealing with disease-modifying therapies, heart failure (HF) specialists also manage the complications of HF. This module will familiarise the reader with the clinical significance and treatment of the more commonly encountered problems: electrolyte disturbances, anaemia and iron deficiency and renal dysfunction.
Electrolyte abnormalities in patients with HF
Electrolyte disturbances are a common complication of HF, which affect treatment decisions and outcomes (table 1).1–7 Hyponatraemia, both hypo- and hyperkalaemia, and more recently, hypochloraemia, are the best-defined electrolyte abnormalities. However, abnormalities in calcium, phosphate, bicarbonate and magnesium may also be common in patients with HF,8 and can be life-threatening if severe, requiring urgent treatment.9
Hyponatraemia
|
Hyponatraemia is the most common electrolyte abnormality seen in patients hospitalised with HF. |
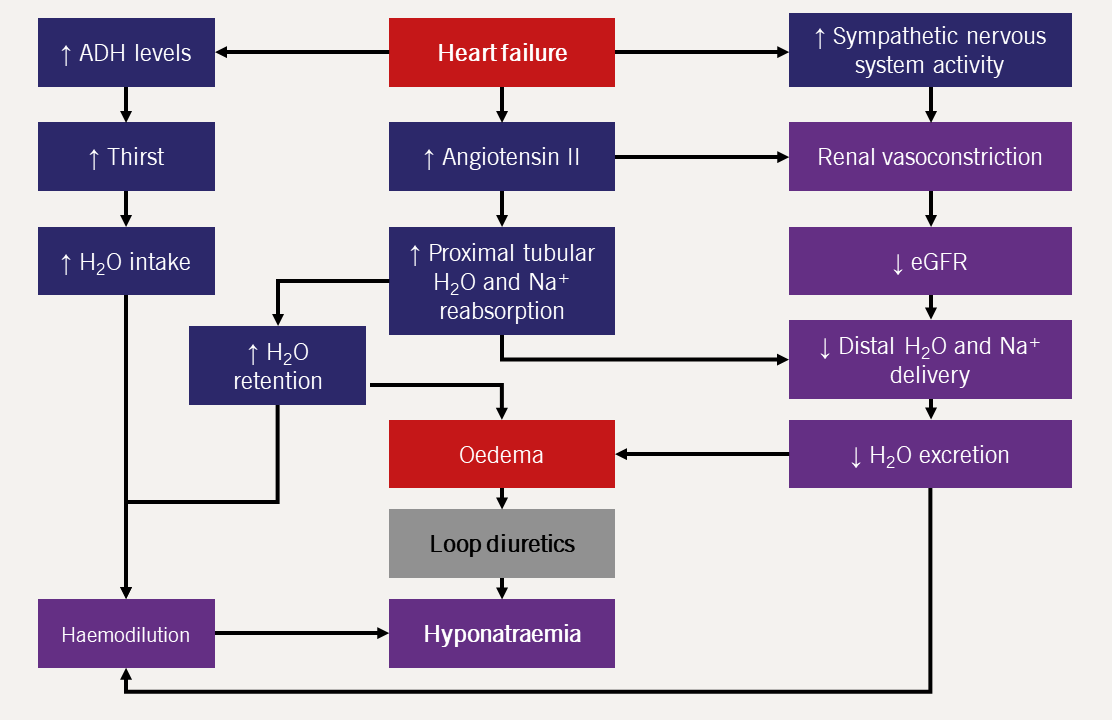
Hyponatraemia is strongly associated with an adverse outcome in acute and chronic HF and is a marker of severe HF that might prompt consideration for transplant.10 It arises through two mechanisms: dilution and/or depletion.11
| Key: ADH = antidiuretic hormone; eGFR = estimated glomerular filtration rate |
- Dilution12
- Depletion
Decreased blood pressure due to HF stimulates baroreceptors in the atria, aorta and peripheral arteries leading to an increase in the activity of the renin-angiotensin-aldosterone and sympathetic nervous systems.
It also causes increased secretion of anti-diuretic hormone (ADH; also known as arginine vasopressin). ADH is normally secreted in response to increasing plasma osmolality, and so in patients with HF, there must be a non-osmolar stimulus to its release.12 The net result is water retention in excess of sodium retention (figure 1).13
Additionally, reduced renal blood flow reduces glomerular filtration rate, thus limiting the rate at which the kidneys can excrete free water.14 Increased water retention leads to dilution of the blood and a fall in sodium concentration, even though the body has an absolute excess of sodium.
The first-line treatment of venous congestion in HF is loop diuretics which act by inhibition of the sodium-potassium-chloride (Na2+-K+-2Cl–) symporter in the ascending limb of the loop of Henlé. The resulting increase in urine sodium, potassium and chloride causes increased free water excretion alongside electrolyte loss.15
However, in the context of low renal perfusion, the ability of the kidneys to produce dilute urine is reduced; under such circumstances, loop diuretics cause urinary electrolyte loss without significant free water excretion.16
Approximately 90% of patients admitted with acute HF are treated with loop diuretics and a similar proportion remains on them one year from discharge.17
Treatment of hyponatraemia
Hyponatraemia is strongly associated with greater mortality in patients with acute or chronic HF.18,19 A patient whose hyponatraemia recovers to normal during an admission for HF has a better outcome than patients with persistent hyponatraemia at discharge.20 However, it is unknown whether treatments which specifically aid to correct low sodium concentrations improve outcomes. Possible treatments include salt restriction, salt supplementation and ADH (arginine vasopressin) receptor antagonists.
- Salt restriction or salt supplementation?
- improve diuresis (measured by reduction in body weight)24
- increase serum sodium24
- cause a greater reduction in natriuretic peptide (NP)25
- reduce length of hospital stay25
- reduce readmission rate.25
- ADH receptor antagonists
Treatment of hyponatraemia in HF remains a controversial subject: in practice, many patients admitted with HF are placed on a fluid restriction in the hope that limiting oral free water intake, in conjunction with loop diuretics, will treat haemodilution. However, evidence to support this is scant and current guidelines recommend fluid restriction to 1.5–2.0 L/day for the treatment of congestion rather than hyponatraemia – again, in the absence of any supporting evidence that this is the correct approach.21 Similarly, sodium restriction is often used, but without supporting evidence. A low-salt diet is unpalatable, and just induces thirst rather than normalising sodium.22
There is some evidence that high-sodium diets23 or sodium supplementation24,25 is the correct approach. Compared to high-dose intravenous (IV) loop diuretic alone, IV hypertonic saline in conjunction with a diuretic in patients admitted with HF and severe fluid retention may:
The dose of diuretic used in these studies was far higher than recommended in guidelines (500–1,000 mg twice daily) and it is possible that the ‘beneficial’ effect of hypertonic saline is merely protective against such high doses of loop diuretics. However, one small study (n=44; New York Heart Association [NYHA] class III–IV; median left ventricular ejection fraction [LVEF], 32%) found greater diuresis (measured by urine volume) and fall in NPs with 40 mg of furosemide in 500 ml of 1.7% saline infused over 24 hours compared to the same dose of diuretic infused over the same time period in 5% glucose.26 More work, as ever, is required.
ADH receptor antagonists can treat hyponatraemia in patients with syndrome of inappropriate secretion of ADH (SIADH). Early trials of ADH receptor antagonists, when added to standard diuretic treatment in patients with HF, showed reductions in patient symptoms and signs of congestion,27 while also increasing serum sodium concentrations more effectively than fluid restriction alone.28 There have been three multi-centre RCTs of ADH receptor antagonists in patients admitted with HF: EVEREST (Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan),27,29 TACTICS-HF (Targeting Acute Congestion with Tolvaptan in Congestive Heart Failure),30 and SECRET of CHF (Study to Evaluate Challenging Responses to Therapy in Congestive Heart Failure).31
The EVEREST trial investigators enrolled 4,133 patients admitted with HF (average age, 66 years; average LVEF, 27.5%).29 Patients were randomised to either tolvaptan 30 mg per day or placebo. The co-primary end points were all-cause mortality and cardiovascular (CV) mortality or HF hospitalisation.29 Treatment with tolvaptan conferred no survival benefit, despite increasing serum sodium among those who were hyponatraemic.29
Subsequent post-hoc analysis amongst patients with hyponatraemia (<135 mmol/L; n=475) found that treatment with tolvaptan was associated with greater weight loss and dyspnoea relief compared to placebo and, amongst patients with severe hyponatraemia (<130 mmol/L; n=92), treatment with tolvaptan was associated with reduction in CV death or hospitalisation (hazard ratio [HR] 0.60; 95% confidence interval [CI], 0.37–0.98; p=0.04).32
The TACTICS-HF trial investigators enrolled 257 patients admitted with HF (average age, 65 years; average LVEF, 33%; average N-terminal of the prohormone of B-type NP [NT-proBNP], 10,246 ng/L; all of whom had serum sodium ≤140 mmol/L at randomisation).30 Patients were randomised to either tolvaptan 30 mg per day or matching placebo alongside fixed-dose IV furosemide.30 The primary end point was a ‘moderate’ improvement in patient-reported breathlessness at eight and 24 hours after starting treatment. Secondary end points included weight and fluid loss at 48 and 72 hours. Treatment with tolvaptan had no impact on the primary end point compared to placebo but was associated with greater weight and fluid loss in the first 48 hours – although there was no significant difference between the two groups after 72 hours.30
The very similar SECRET of CHF trial enrolled 250 patients admitted with HF (average age, 70 years; average LVEF, 35%; median B-type NP [BNP], 577 ng/L in the treatment arm) who either had renal impairment (estimated glomerular filtration rate [eGFR] <60 ml/min/1.73 m2), hyponatraemia (≤134 mmol/L) or diuretic resistance.31 Patients were randomised to either tolvaptan 30 mg per day or matching placebo. The primary end point was an improvement in patient-assessed breathlessness after 24 hours of treatment with weight and net fluid loss amongst the pre-specified secondary end points.31 Again, treatment with tolvaptan had no effect on symptoms, but was associated with greater weight loss compared to treatment with furosemide alone.31 In a pre-specified sub-group analysis amongst patients with hyponatraemia, similar effects were observed, regardless of the presence of hyponatraemia.32
Table 2. Diuretic effects of tolvaptan in addition to intravenous furosemide in the EVEREST, TACTICS-HF and SECRET of CHF trials
| Trial | N | Mean daily furosemide dose | Daily tolvaptan dose | Diuretic effect | |
| EVEREST29,32 | 4,133 | 235 mg | 30 mg | Change in body weight from baseline | |
| Day 1 p<0.001 |
Tolvaptan −1.76 kg | ||||
| Placebo −0.97 kg | |||||
| TACTICS-HF30 | 257 | 71 mg | 30 mg | Change in body weight from baseline | |
| After day 1 p=0.005 |
Tolvaptan −2.0 kg | ||||
| Placebo −0.5 kg | |||||
| After day 2 p=0.004 |
Tolvaptan −2.8 kg | ||||
| Placebo −1.6 kg | |||||
| After day 3 p=0.07 |
Tolvaptan −3.7 kg | ||||
| Placebo −2.5 kg | |||||
| Net fluid loss | |||||
| On day 1 p=0.006 |
Tolvaptan −2182 ml | ||||
| Placebo −1541 ml | |||||
| On day 2 p=0.01 |
Tolvaptan −1948 ml | ||||
| Placebo −1419 ml | |||||
| On day 3 p=0.11 |
Tolvaptan −1757 ml | ||||
| Placebo −1401 ml | |||||
| SECRET of CHF31 | 250 | 160 mg | 30 mg | Change in body weight from baseline | |
| After day 1 p<0.001 |
Tolvaptan −2.4 kg | ||||
| Placebo −0.9 kg | |||||
| After day 2 p<0.001 |
Tolvaptan −3.1 kg | ||||
| Placebo −1.9 kg | |||||
| After day 3 p=0.006 |
Tolvaptan −3.6 kg | ||||
| Placebo −2.4 kg | |||||
| Key: EVEREST = Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan; SECRET of CHF = Study to Evaluate Challenging Responses to Therapy in Congestive Heart Failure; TACTICS = Targeting Acute Congestion with Tolvaptan in Congestive Heart Failure Study | |||||
Taken together, the data suggest that vasopressin antagonists may be useful adjuncts to loop diuretics to stimulate a diuresis in cases of diuretic resistance, regardless of serum sodium. At present, they are only ‘suggested’ for the treatment of resistant hyponatraemia in the context of congestion.21
Hypokalaemia
Box 1. Electrocardiogram changes associated with hypokalaemia
| U waves T-wave flattening ST-segment changes Ventricular tachycardia Pulseless electrical activity or asystole |
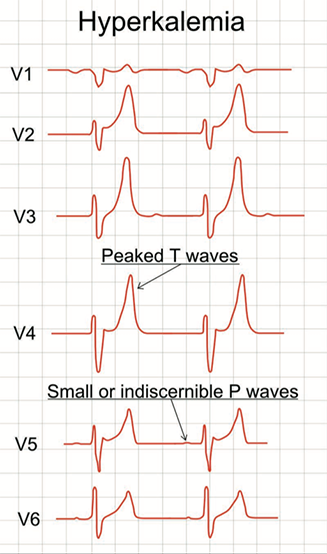
Hypokalaemia in patients with HF is most often a consequence of diuretic therapy.33 Hypokalaemia prolongs ventricular repolarisation, slows conduction between myocytes and reduces intrinsic pacemaker activity; all of which predispose to atrial and ventricular arrhythmias.34 In animal studies, even moderate hypokalaemia (2.5–3.0 mmol/L) can be highly arrhythmogenic; 50% of the rabbits tested had either ventricular tachycardia (VT) or ventricular fibrillation (VF) after reducing serum potassium to 2.7 mmol/L.35 Electrocardiogram (ECG) changes due to hypokalaemia indicate those at greatest risk of arrhythmia (box 1) (figure 2).
| Reproduced with the kind permission of Han B Xiao ©Concise Clinical Consulting Ltd. |
Treatment of hypokalaemia
Correction of hypokalaemia is indicated if ECG changes are present or if potassium is <2.5 mmol/L.9 Unless cardiac arrest is imminent, potassium replacement should be at a rate of 10–20 mmol/hr given intravenously with sodium chloride 0.9% solution. Oral supplementation may be sufficient in mild cases (3.0–3.4 mmol/L).
Hypomagnesaemia increases renal excretion of potassium and is often seen in conjunction with hypokalaemia; thus, correction of concurrent hypomagnesaemia is essential for effective potassium replacement.36
Of course, treatment with mineralocorticoid receptor antagonists (MRAs) is the most obvious long-term option for either treatment or prevention of hypokalaemia in patients with HF due to reduced ejection fraction (HFrEF) – which may be one mechanism by which the medication provides prognostic benefit.
Hyperkalaemia
Some 90% of potassium excretion is via the kidneys. Once filtered through the glomerulus, the majority is then reabsorbed from the urine in the proximal convoluted tubule and loop of Henlé.37 Excretion of excess serum potassium into the urine occurs via Na+/K+-ATPase pumps in the principal cells of the collecting duct which reabsorb sodium in exchange for potassium ions. The pumps are upregulated by aldosterone.38,39 Thus, a reduction in serum aldosterone (or mineralocorticoid receptor antagonism with spironolactone or eplerenone) reduces urinary excretion of potassium.40
Hyperkalaemia (>6.0 mmol/L) is associated with higher mortality in patients with HF5 and discontinuation of drugs with proven prognostic benefit (such as MRAs) to avoid iatrogenic hyperkalaemia is common.41
Causes of hyperkalaemia in patients with HF
- Renin-angiotensin-aldosterone system (RAAS) inhibitors
- MRAs
- Concomitant conditions: Diabetes and chronic kidney disease (CKD) are associated with hyperkalaemia (>5.5 mmol/L), independent of treatment with an MRA.50,51
RAAS inhibitors are associated with higher serum potassium and increased risk of hyperkalaemia (figure 3).42,43
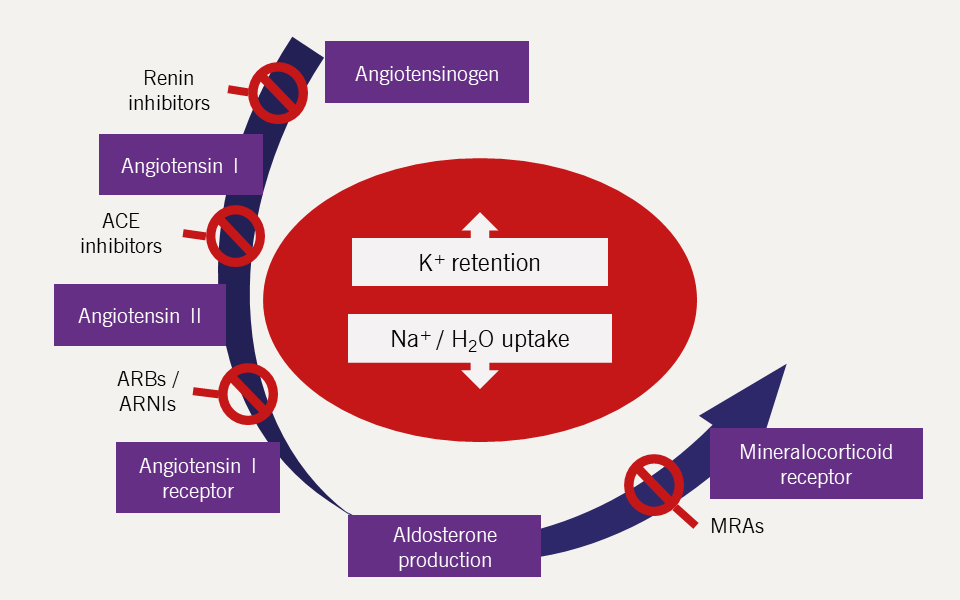
| Key: ACE = angiotensin-converting enzyme; ARB = angiotensin II receptor blocker; ARNI = angiotensin receptor–neprilysin inhibitor; MRA = mineralocorticoid receptor antagonist; |
The rate of hyperkalaemia is higher with MRAs than with angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) (table 3).44–47 Perhaps as few as 54% of cases of hyperkalaemia among patients with HF who are taking an MRA are attributable to the drug itself.49
Table 3. Rates of hyperkalaemia in clinical trials of RAAS inhibitors in patients with HF
| Trial | Drug | Follow-up duration | Hyperkalaemia definition | Rates of hyperkalaemia | |
| Treatment | Control | ||||
| SOLVD44 (1991) | Enalapril | 41 months | ≥5.5 mmol/L | 7.8% | 4.2% |
| CHARM-Added45 | Candesartan | 41 months | Hyperkalaemia associated with an adverse event† | 8.4% | 2.9% |
| RALES46 (1999) | Spironolactone | 24 months | ≥5.5 mmol/L | 19.0% | 5.6% |
| EMPHASIS-HF47 (2011) | Eplerenone | 21 months | ≥5.5 mmol/L | 5.4% | 3.8% |
| PARADIGM-HF48 (2014) | Sacubitril/valsartan | 27 months | ≥5.5 mmol/L | 16.1% | 17.3% |
| † Death, dose reduction or drug discontinuation for any reason Key: CHARM-Added = Candesartan in Heart Failure – Assessment of Mortality and Morbidity; EMPHASIS-HF = Eplerenone in Patients with Systolic Heart Failure and Mild Symptoms; PARADIGM-HF = Prospective Comparison of ARNI [Angiotensin Receptor–Neprilysin Inhibitor] with ACEI [Angiotensin-Converting–Enzyme Inhibitor] to Determine Impact on Global Mortality and Morbidity in Heart Failure; RAAS = renin-angiotensin-aldosterone system; RALES = Effect of Spironolactone on Morbidity and Mortality in Patients with Severe Heart Failure; SOLVD = Studies of Left Ventricular Dysfunction |
|||||
Combination therapy can thus be dangerous, making monitoring of potassium mandatory in patients receiving both an ACE inhibitors/ARBs and an MRA.21,52 In clinical trials, however, hyperkalaemia is also seen in the placebo groups, and thus hyperkalaemia is not solely due to HF treatment.
Box 2. Electrocardiogram changes associated with hyperkalaemia
| Peaked T-waves (‘tall, tented T-waves’) Broad, small or absent P-waves Broad QRS complexes Varying degrees of heart block Asystole Sine-wave pattern – T-waves fused with a broad QRS complexes Idioventricular rhythm Ventricular tachycardia / fibrillation |
As with hypokalaemia, hyperkalaemia is associated with an increased risk of tachy- and bradyarrhythmias. High intracellular potassium causes shortening of the action potential duration and slowed conduction velocity of the action potential across myocytes.34 The consequence is impaired atrioventricular (AV) node conduction and heart block which – in conjunction with suppression of infranodal pacemaking (the ability of ventricular myocardium to generate an ectopic beat) in hyperkalaemia – may cause asystole.34 Hyperkalaemia also predisposes to VT/VF through an incompletely understood mechanism.38
The appearance of ECG changes with hyperkalaemia (box 2, figure 4) is a medical emergency which requires urgent treatment (see below).9
| Reproduced from Mikael Häggström, Public domain, via Wikimedia Commons https://commons.wikimedia.org/wiki/File:ECG_in_hyperkalemia.svg |
Treatment of hyperkalaemia
Moderate to severe hyperkalaemia
The goal of emergency treatment of hyperkalaemia is to treat/prevent the associated arrhythmias and is generally only required for patients with serum potassium levels >6.0 mmol/L (figure 5).34
Treatment options
- Potassium binders
- Patiromer calcium
- ZS-9
Patiromer calcium is recommended by the National Institute for Health and Care Excellence (NICE) as an option for treating patients with persistent hyperkalaemia (≥6.0 mmol/L) and HF, if they are not taking, or are taking a reduced dosage of a RAAS inhibitor because of hyperkalaemia and are not on dialysis.53
NICE currently recommends sodium zirconium cyclosilicate (ZS-9) for the emergency treatment of life-threatening hyperkalaemia or in outpatients with either HF or CKD stage IIIb–V with persistently high serum potassium levels (≥6.0 mmol/L) who are not on dialysis and are not taking an ‘optimised dose’ of RAAS inhibitor due to hyperkalaemia.54
The guidance is couched in very cautious terms, recognising the weakness of the available evidence for long-term use.
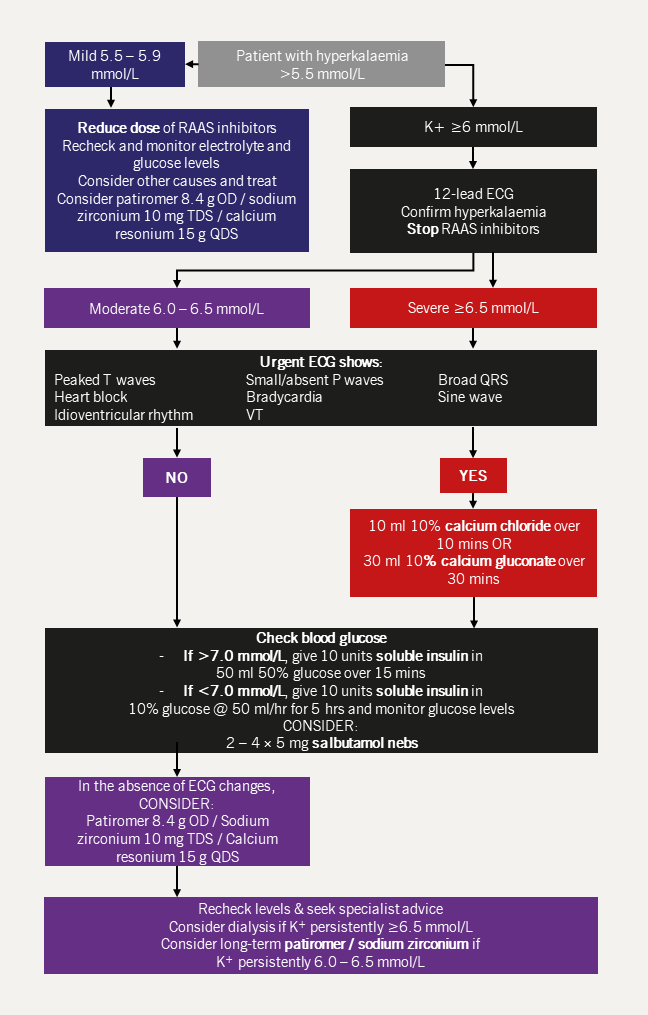
| Key: ECG = electrocardiogram; RAAS = renin-angiotensin-aldosterone system; OD = once daily; QDS = quater die sumendum (four times daily); TDS = ter die sumendum (three times daily); VT = ventricular tachycardia |
Evidence
Prior to the advent of the newer potassium binders, the only treatment available (beyond emergency measures) was calcium resonium: a polymer derived from polystyrene which bound to potassium ions in the gastrointestinal (GI) tract, thus increasing excretion. However, the polymer swells when in contact with water and GI side effects are common, making it unsuitable for longer term use.55 Thus clinicians and patients had few options when presented with the problem of hyperkalaemia due to treatment with ACE inhibitor, ARB or MRA: reduce the dose or stop altogether.
Newer potassium binders are non-absorbable compounds that bind potassium in the gut without such high rates of GI side effects. Two drugs have been investigated for the treatment and prevention of hyperkalaemia in patients with HF: patiromer and sodium zirconium cyclosilicate (ZS-9); both have received technology appraisal guidance from NICE,53,54 but it is unlikely either will be commonly used in routine practice.
At least part of the benefit of MRAs can be attributed to prevention of hypokalaemia; for example, patients with HF who take potassium-sparing diuretics have a lower risk of adverse outcomes compared to those taking non-potassium-sparing diuretics.56 Furthermore, potassium concentration of 5.0–5.5 mmol/L are associated with the lowest risk of adverse outcomes in patients with chronic HF.5 In a patient who has to stop or reduce MRA for hyperkalaemia, there is no evidence that taking both an MRA and a potassium binder is better than taking neither.
In the PEARL-HF trial (Prevention of Hyperkalemia in Patients with Heart Failure Using a Novel Polymeric Potassium Binder, RLY5016), investigators enrolled 105 patients with HF (average age, 68 years; average LVEF, 40%), all of whom had previously had an ACE inhibitor, ARB or MRA discontinued for hyperkalaemia or had CKD.57 Patients were randomised to either patiromer 30 g per day or matching placebo.57 All patients (except two in the placebo arm) were taking an ACE inhibitor or an ARB and all patients started spironolactone 25 mg on the same day as the study drug.57
Treatment with patiromer (30 g per day for four weeks) was associated with a lower rate of hyperkalaemia (≥5.5 mmol/L) compared to placebo (7% vs. 25%; p=0.015).57 This allowed the dose of spironolactone to be increased (from 25 mg to 50 mg once daily) in a higher proportion of patients taking patiromer compared with placebo (91% vs. 74%; p=0.019).57
Adverse event rates were low; GI disturbances such as flatulence, diarrhoea and constipation were the most commonly reported in the patiromer group, but only 7% of patients required discontinuation of the study drug. A subsequent smaller trial (n=63) found that treatment with patiromer enabled patients who would otherwise be classed as high risk of developing hyperkalaemia (CKD and serum potassium 4.3–5.1 mmol/L) to achieve target dose of spironolactone (50 mg once daily).58
The largest trial of patiromer to date – the DIAMOND trial (Patiromer for the Management of Hyperkalemia in Subjects Receiving RAAS Inhibitor Medications for the Treatment of Heart Failure) – enrolled 1,195 patients with HFrEF and a history of hyperkalaemia with RAAS inhibitor treatment (average age, 66 years; 25% women; 50% NYHA class II; about 45% of whom had an eGFR <60 ml/min/1.73 m2, and about 40% of whom had serum potassium concentrations >5.0 mmol/L at screening).59 DIAMOND was originally designed to assess the effect of long-term patiromer on CV hospitalisation or death, but due to slow recruitment, the end point was altered to change in serum potassium from baseline.
Patiromer was associated with a 0.1 mmol/L reduction in serum potassium. Hyperkalaemia (>5.5 mmol/L) occurred in 13.9% of patients in the patiromer group and 19.4% of patients in the control arm (p=0.006).59 Hypokalaemia was just as common as hyperkalaemia and more likely to occur in the patiromer arm (15% vs. 10.7%). Rates of MRA discontinuation were low in both arms with no difference between the groups.59
The primary findings of the DIAMOND trial are that 1) hyperkalaemia is an uncommon side effect in patients with HFrEF taking RAAS inhibitor medications, even in patients with a history of hyperkalaemia on RAAS inhibitors; and 2) patiromer has a statistically significant but clinically unimportant effect on serum potassium concentrations.59
In the HARMONIZE trial (Effect of Sodium Zirconium Cyclosilicate on Potassium Lowering for 28 Days Among Outpatients with Hyperkalemia) of ZS-9 in patients with hyperkalaemia regardless of aetiology, investigators enrolled 258 patients with potassium ≥5.1 mmol/L and randomised them to either 5 g, 10 g or 15 g of ZS-9 per day or matching placebo for 28 days.60 The proportion of patients with potassium <5.1 mmol/L was significantly greater in all ZS-9 groups compared to placebo (p<0.001).60 The effect of ZS-9 was rapid; during the initial open-label phase (during which all patients received the drug), 98% of patients treated with ZS-9 had normal potassium with the median time to normalisation of only about two hours.60
Subgroup analysis of patients with HF in the HARMONIZE study (n=94; 70% had CKD or diabetes; 69% taking ACE inhibitors, ARBs or MRAs) found similar rapid and beneficial improvements in serum potassium with ZS-9 versus placebo.61
There were no serious adverse events or events that led to study drug discontinuation during the open-label phase. The adverse event rate was similar between the ZS-9 and placebo groups during the maintenance phase. The most common adverse event in the ZS-9 group was peripheral oedema in patients taking either 10 g per day (6%) or 15 g per day (14%), the clinical significance of which is not clear with such small numbers.60
In a combined analysis of patients with severe hyperkalaemia (≥6.0 mmol/L, n=45) from two studies of ZS-9, investigators found that 80% of patients achieved potassium levels <6.0 mmol/L within four hours of starting treatment5 and thus ZS-9 may also be helpful for the emergency management of severe hyperkalaemia.62
The HARMONIZE-global study (n=267; average age, 67 years; ~20% with HF; 98% with risk factors of hyperkalaemia – CKD, diabetes, HF or RAAS inhibitor use) aimed to replicate the results of the HARMONIZE trial in a more ethnically diverse group of patients with hyperkalaemia (≥5.1 mmol/L).63 In the initial 48-hour ‘correction phase’ of the study, all patients received 10 g ZS-9 three times per day. Those who reached normal potassium levels (<5.1 mmol/L, n=248, 92.9%) were then entered into the 29-day ‘maintenance phase’ in which patients were randomised to either once-daily ZS-9 5 g, ZS-9 10 g or matching placebo in a 2:2:1 ratio.63
The primary end point was average serum potassium during days 8–29 of the maintenance phase of the trial.63 Potassium fell to within the study-defined normal range (<5.1 mmol/L) in both ZS-9 groups more frequently than in the placebo group (p<0.001).63 At the end of the study, 77.3% of patients taking ZS-9 10 g per day and 58.6% of patients taking ZS-9 5 g per day had normal potassium compared to only 24.0% of patients taking placebo (p<0.001).63 Interestingly, serum aldosterone levels were significantly lower in patients taking ZS-9 compared to placebo at the end of the study compared to baseline (p=0.001).63
While the adverse event rate was high (44.4% in the ZS-9 10 g/day group and 28.3% in the ZS-9 5 g/day compared to 20.0% in the placebo group), the rate of discontinuation of the study drug was low and similar between the groups (7% vs. 7% vs. 6%).63 The most commonly reported adverse event associated with ZS-9 was new-onset oedema occurring in 15% and 5% of patients taking 10 g and 5 g, respectively.63 While the exact mechanism by which ZS-9 may cause oedema is unknown, by the end of the study, oedema had resolved or was resolving in most patients (80%) either without specific treatment or with loop diuretic without requiring discontinuation of the study drug.63
Renal dysfunction in patients with HF
Renal dysfunction is common in patients with HF;64,65 while one in five patients with HF have a label of CKD, as many as 50% of patients may have an eGFR <60 ml/min/1.73 m2.66
Renal dysfunction may be a consequence of HF, its treatment, or both, and is invariably associated with a poor prognosis in trial and registry data, regardless of HF phenotype.67–70
In clinical practice, there is frequently a tension between the need to initiate and titrate drugs that inhibit the RAAS on the one hand, and the avoidance of further detriment to renal function on the other.
The inclusion of ACE inhibitors, ARBs and MRAs in lists of ‘nephrotoxic’ drugs, combined with unclear, and occasionally, conflicting clinical guidelines have resulted in the sometimes ‘hair-trigger’ response of stopping an ACE inhibitor, ARB or MRA in response to any deterioration in renal function without any attempt to consider an alternative cause or reintroduce the medications in the future.
The situation is compounded by the frequently conflicting recommendations given to GPs by nephrologists, cardiologists and other hospital specialists.
Causes of renal dysfunction in HF12
The cause of renal dysfunction in patients with HF is multifactorial. Additionally, patients with HF are vulnerable to acute intercurrent illnesses, which are a common cause of a decline in renal function. Intercurrent illness must always be considered in a patient with HF who is taking RAAS inhibitors and who had previously stable renal function. The commonest factors are:
- Haemodynamic and neurohormonal changes12
- Venous congestion
- Atherosclerosis
- Effect of treatments
Glomerular filtration is normally maintained by the action of the RAAS across a wide range of systolic blood pressures. The low-flow state of HF causes increased dependency on activation of the RAAS to maintain glomerular filtration pressure.
Angiotensin II causes vasoconstriction of the afferent (in) and efferent (out) arterioles of the glomerulus. Vasoconstriction is more pronounced in the efferent arteriole, causing increased flow into the glomerulus via the afferent arteriole (less vasoconstriction – larger relative vessel diameter) and decreased flow out via the efferent arteriole (more vasoconstriction – smaller relative vessel diameter). The net effect is increased pressure within the glomerulus, which drives filtration from the glomerulus to the Bowman’s capsule and into the urinary space.
Therefore, introduction of ACE inhibitors, ARBs, MRAs and sacubitril/valsartan inevitably cause a reduction in glomerular filtration pressure on initiation and cause apparent deterioration in renal function tests.71 With inhibition of the RAAS, glomerular filtration becomes more dependent on systolic blood pressure to perfuse the glomerulus adequately. Blood pressure is, of course, reduced by the systemic effects of ACE inhibitors, ARBs, MRAs or sacubitril/valsartan (alongside beta blockers).72 Thus, a degree of renal impairment after initiation of inhibitors of the RAAS is almost ubiquitous.
Renal interstitial oedema increases pressure in the Bowman’s capsule, thus reducing the filtration pressure from glomerulus to Bowman’s capsule and reducing the filtration rate. Similarly, high pressure in the renal vein lowers eGFR and increases sodium retention, although the mechanism is unclear.73 Additionally, patients with severe congestion can develop ascites, which may cause functional ureteric obstruction due to increased intra-abdominal pressure.74
Coronary artery disease is common amongst patients with HF and is often associated with other arteriopathies, such as peripheral vascular disease or renal artery stenosis.12 The prevalence of renal artery stenosis in patients with HF varies from 15–54%, depending on the definition used and the population studied.75,76 A stenosed renal artery reduces renal blood flow, in turn leading to renin production as a mechanism to maintain glomerular perfusion. Inhibition of the RAAS in such patients can lead to a profound drop in renal function, which is not always reversible and may lead to life-threatening pulmonary oedema.77
- RAAS inhibitor trials
- In the CONSENSUS trial (Effects of Enalapril on Mortality in Severe Congestive Heart Failure), the average increase in serum creatinine in patients treated with enalapril was 44 µmol/L. Creatinine decreased in about 25% of patients in the treatment group78,79
- In the SOLVD Prevention trial (Studies of Left Ventricular Dysfunction), the proportion of patients with serum creatinine increase >44 µmol/L was 16% in the enalapril arm compared to 12% in the placebo arm80
- In RALES (Randomized Aldactone Evaluation Study), the median increase in creatinine in the spironolactone group was 4–9 µmol/L46
- In the PARADIGM-HF study (Prospective Comparison of ARNI With ACE inhibitor to Determine Impact on Global Mortality and Morbidity in Heart Failure), the proportion of patients who developed creatinine ≥221 µmol/L was 3% in the sacubitril/valsartan group and 5% in the enalapril group.48
- Diuretics
RAAS inhibitors – Trial data suggests that while a large proportion of patients will experience a decline in renal function on starting RAAS inhibitors, the change is modest
In a post-hoc analysis of both SOLVD and RALES, worsening renal failure upon starting treatment was associated with poorer prognosis amongst patients in the placebo group but not the treatment group.81,82
Similarly, a meta-analysis of data from the SOLVD (enalapril), RALES (spironolactone), SAVE (Survival and Ventricular Enlargement — captopril), Val-HeFT (Valsartan Heart Failure Trial — valsartan) and EPHESUS (Eplerenone, a Selective Aldosterone Blocker, in Patients with Left Ventricular Dysfunction After Myocardial Infarction — eplerenone) trials found that while worsening renal function was associated with higher mortality compared to patients with stable renal function.83 Additionally, the overall reduction in all-cause mortality associated with RAAS inhibitors was greater amongst patients with worsening renal function compared to those with stable renal function.83
Diuretics – Diuretics are the cornerstone of treatment for venous congestion but are often associated with worsening renal function. Although worsening renal function is often associated with worse outcome,84 it does not demonstrate causality.
Post-hoc analysis of the DOSE trial (Diuretic Strategies in Patients with Acute Decompensated Heart Failure) (module 3) suggests that worsening renal function during diuresis may actually be associated with better post-discharge outcomes compared to patients whose renal function is stable or improves.85
In a study of approximately 600 consecutive patients admitted with acute HF, the presence of worsening renal failure on discharge (≥ 26.5 µmol/L increase in serum creatinine from admission) was not associated with adverse outcomes, whereas the presence of residual congestion was.86 Ultimately, treatment of congestion – not maintenance of stable renal function – is the goal of treatment during diuresis, and the two rarely go hand-in-hand.
Thresholds for changing medication
|
The European Society of Cardiology (ESC) HF guidelines permit a creatinine increase of ≤50% from baseline before recommending dose reduction or withdrawal. |
The decision to reduce the dose or withdraw the treatment with a RAAS inhibitor is based on a risk–benefit calculation, for which there is very little evidence on which to base a decision. Current NICE HF guidelines refer to NICE CKD guidelines, which recommend either dose reduction or withdrawal of ACE inhibitors, ARBs or MRAs if serum creatinine rises >30% from baseline.87
What action is taken depends very much on the clinical context and the indication for which the medication was prescribed. For example, there is no evidence that treatment with RAAS inhibitors improves outcomes for patients with HF and a preserved left ventricular ejection fraction (HFpEF) and thus the threshold to reduce the dose or withdraw treatment altogether is much lower than for patients with HFrEF in whom the RAAS inhibitors unequivocally confer prognostic benefit.88
The British Society for Heart Failure and the Renal Association released guidelines on how to manage deteriorating renal function and hyperkalaemia in patients with HF (table 4).88
Table 4. Recommendations from the RA and BSH regarding changes in medication in response to changes in renal function
| Increase in serum creatinine from baseline | HFpEF | HFrEF |
| ≤30% | Consider stopping RAAS inhibitor | Continue |
| 30–50% | Stop RAAS inhibitor | Consider reduced dose or temporary withdrawal |
| >50% | Temporary withdrawal | |
| eGFR <20 ml/min/1.73 m2 | Stop RAAS inhibitor | |
| Key: BSH = British Society for Heart Failure; eGFR = estimated glomerular filtration rate; HFpEF = heart failure with preserved ejection fraction; HFrEF = heart failure with reduced ejection fraction; RA = Renal Association; RAAS = renin-angiotensin-aldosterone system | ||
The recommendations mean that in a patient with HFrEF, an increase in creatinine from 150 µmol/L at baseline to 190 µmol/L is tolerable, as long as it is not accompanied by another reason for dose adjustment, such as hyperkalaemia ≥6.0 mmol/L or symptomatic hypotension.88 However, such an increase in a patient with HFpEF should prompt dose reduction or withdrawal.88
There is a danger that anxiety about renal function may lead to underuse of potentially life-prolonging medications in patients with HF.89 However, real-life clinical scenarios are rarely as simple as the arbitrary cut-offs described above may imply and ultimately, the decision to reduce the dose of or stop treatment with RAAS inhibitors will be based on other factors as well as serum creatinine.
Anaemia in patients with HF
Anaemia is common in patients with HF. It is most often seen in women, the elderly and patients with renal dysfunction.90 It is associated with more severe symptoms, worse functional status, and greater morbidity and mortality.91,92 The aetiology is multifactorial.
The prevalence of anaemia varies from 7–50% in study populations and HF registries due to varying definitions and patient selection for studies (figure 6). In a systematic review of 34 studies, most of which used the World Health Organisation definition of anaemia (haemoglobin [Hb] <13 g/dL for men; <12 g/dL for women), the prevalence of anaemia was 37% amongst 153,000 patients with HF.93
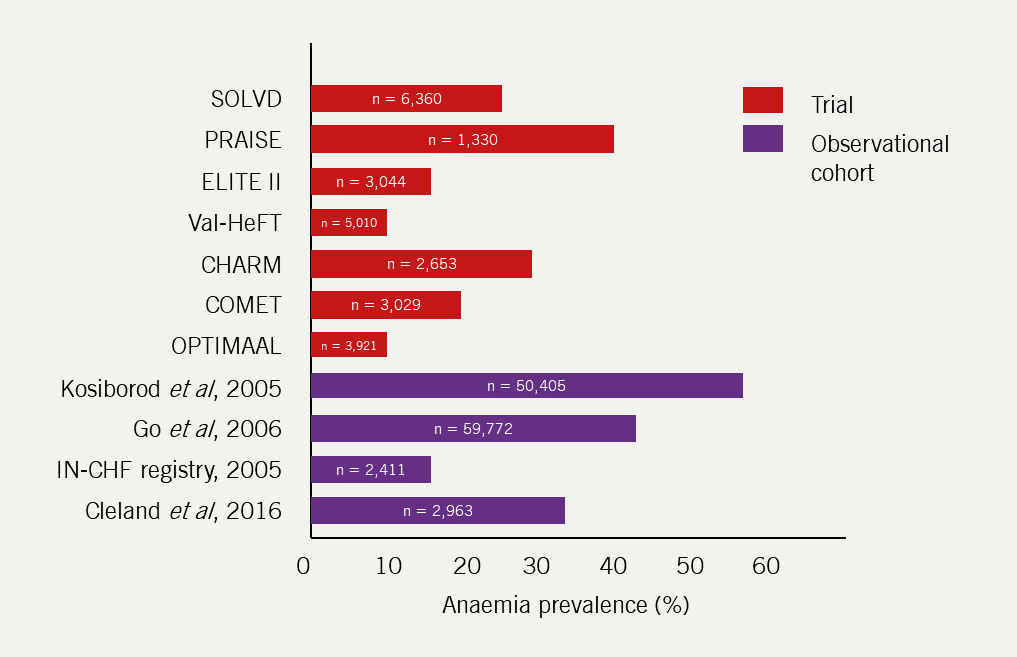
| Key: CHARM = Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity; COMET = Carvedilol Or Metoprolol European Trial; ELITE = Evaluation of Losartan in the Elderly; IN-CHF = Italian Network on Congestive Heart Failure; OPTIMAAL = Optimal Therapy in Myocardial Infarction with the Angiotensin II Antagonist Losartan; PRAISE = Prospective Randomized Amlodipine Survival Evaluation; SOLVD = Studies of Left Ventricular Dysfunction; Val-HeFT = Valsartan Heart Failure Trial |
The most studied and best understood cause of anaemia in patients with HF is iron deficiency. While the true cause is unclear, it is likely that iron deficiency in patients with HF stems from a combination of reduced iron stores, inability to use intracellular iron stores, poor absorption and sub-acute blood loss (due to oral antiplatelets and anticoagulants).94,95
Antiplatelet agents, such as aspirin, are almost ubiquitous in the treatment of ischaemic heart disease,96 which is the leading cause of chronic HF.65,66 Atrial fibrillation is another common co-morbidity in patients with HF, treatment of which in a patient with HF should involve anticoagulation with either warfarin or direct-acting oral anticoagulants in the absence of a contraindication.12
|
In the general population, iron deficiency is approximately three times more common than iron-deficiency anaemia. Anaemia should be considered the end point of iron deficiency. |
Iron has many physiological roles that may be negatively affected by low serum levels. Physiological roles of iron include99,100:
- forming the oxygen-binding component in both Hb and myoglobin; the latter allows oxygen release in skeletal muscle in hypoxic conditions
- a role in maturation of haematopoetic stem cells
- a role in the cellular response to erythropoietin (EPO)
- the electron emitted by the conversion of Fe2+ to Fe3+ is used as a catalyst for various biochemical reactions and in the electron transport chain in mitochondria.
Patients with HF and iron deficiency have lower six-minute walk test (6MWT) distances and peak exercise oxygen consumption (VO2), and worse outcomes than those with normal iron stores, regardless of the presence or absence of anaemia.91,101,102 While iron deficiency is common in patients with HF, other causes should also be considered.
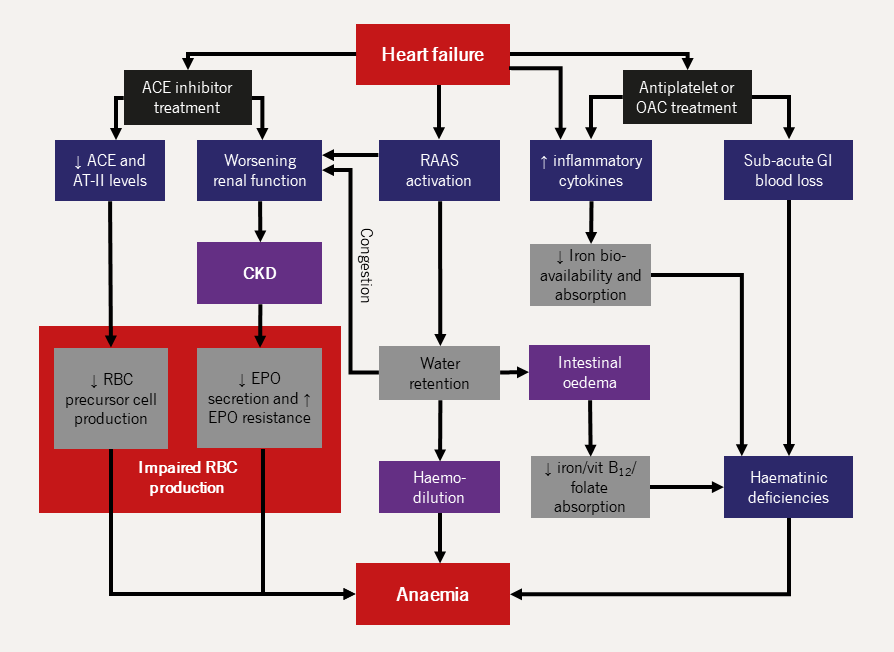
| Key: ACE = angiotensin-converting enzyme; AT-II = angiotensin II receptor type 2; CKD = chronic kidney disease; EPO = erythropoietin; GI = gastrointestinal; OAC = oral anticoagulant; RAAS = renin-angiotensin-aldosterone system; RBC = red blood cell |
Causes of anaemia in HF
- CKD
- Suppression of angiotensin II activity
- Reduced angiotensin II activity causes dilation of the efferent arteriole of the glomerulus and increased renal blood flow which, in turn, reduces EPO secretion
- Angiotensin II stimulates production of red blood cell precursor cells via activation of angiotensin II receptor type 1 on blast cells105
- ACE inhibitors inhibit the breakdown of N-acetyl-seryl-aspartyl-lysyl-proline, which suppresses haemopoetic stem cell proliferation.106
- Effect of pro-inflammatory cytokines
- Venous congestion
- Other haematinic deficiencies
|
CKD is common among patients with chronic HF (see above).65,66 The primary cause of anaemia in patients with CKD is reduced EPO production but other factors, such as those described above, may also play a role. The prevalence of anaemia among patients with CKD is inversely proportional to eGFR (table 5).103 |
Table 5. Prevalence of anaemia by glomerular filtration rate in outpatients with chronic kidney disease (n=18,474)66
|
||||||||||||||
ACE inhibitors may contribute to anaemia in patients with HF as follows:104
HF is a pro-inflammatory state associated with raised tumour necrosis factor and interleukin-6, among other cytokines.107 Levels of these cytokines are inversely proportional to Hb.108 Pro-inflammatory states reduce EPO secretion,109 and reduce GI absorption and the bioavailability of iron for haem production.110
Neurohormonal activation in HF leads to increased salt and water retention, which increases plasma volume. Increased plasma volume in turn can dilute serum components, including Hb. Haemodilution may account for as many as half the cases of anaemia in patients with chronic HF111 and is associated with low Hb in patients with HF, regardless of eGFR and EPO.112
Malnutrition is increasingly recognised as a complication of HF, possibly due to impaired gut absorption secondary to bowel oedema.113,114 However, while malnutrition is a common cause of anaemia worldwide,115 vitamin B12 and folate deficiencies are uncommon in patients with HF.91,116–118 Iron deficiency is far more common: in one international cohort of patients with chronic HF (n=610), 58% had iron deficiency, whereas only 5% and 4% had vitamin B12 and folate deficiencies, respectively.117
Investigating anaemia in patients with HF
|
Most patients with HF are elderly and have multiple co-morbidities and risk factors for malignancy, so those with iron deficiency ought to be offered GI evaluation.114 |
All patients with anaemia (regardless of the presence of HF) will initially require further investigation with haematinics and a blood film.
Current British Society of Gastroenterology guidelines recommend urgent referral for investigation of possible GI malignancy in all patients aged over 60 years with iron-deficiency anaemia (Hb <13 g/dL for men, <12 g/dL for women; ferritin <15 ng/ml) and specialist referral for all patients with iron deficiency and significant anaemia (Hb <12 g/dL for men, <10 g/dL for women), regardless of age.114 However, a large proportion of patients with iron-deficiency anaemia may be sub-optimally investigated.119,120
Whether patients with iron deficiency but normal Hb should be investigated is not clear. Current guidelines recommend coeliac screening in all patients with non-anaemic iron deficiency and consideration of GI evaluation in all patients thought to be ‘high risk’ of malignancy, for example those over 50 years, which of course, includes most patients with HF!114
Treating anaemia in patients with HF
Blood transfusions are the most simple and obvious treatment for anaemia, yet very few data exist that suggests benefit. A blood transfusion, given with a diuretic, may be indicated for patients in whom severe anaemia or a sudden drop in Hb is thought to have caused worsening HF symptoms.
Blood transfusions are recommended for patients with acute coronary syndrome and an Hb ≤8 g/dL to target an Hb 8–10 g/dL; in practice, such targets also seem reasonable for patients hospitalised with HF if anaemia is thought to contribute to worsening symptoms. However, such decisions are purely clinical and have no evidence base to support them.
Of the various aetiologies of anaemia in patients with HF explored above, two mechanisms have been targeted in therapeutic trials to varying degrees of success: iron depletion and low EPO.
Early studies suggested that treatment with erythropoiesis-stimulating agents (ESAs),121 IV iron,122 or both,123 may improve functional capacity in patients with HF and anaemia; large-scale randomised controlled trials (RCT) have followed.
There is no evidence that oral iron or ESAs are beneficial and, despite the adverse safety signals in RED-HF (Reduction of Events by Darbepoetin Alfa in HF),124 the latter is often given to patients with HF and end-stage renal failure. IV iron is recommended by the ESC HF guidelines for the treatment of iron deficiency defined as serum ferritin concentrations <100 μmol/L or ferritin 100–299 and transferrin saturation <20% to improve symptoms and reduce the risk of hospitalisation with HF.21 Screening for iron deficiency at regular intervals is also recommended.
- ESAs
- IV iron
- Oral iron
In the RED-HF trial (Reduction of Events with Darbepoetin Alfa in Systolic Heart Failure) 2,278 patients (median age, 72 years; median LVEF, 31%; median Hb, 11.2 g/dL; median transferrin saturation, 24%)124 were randomised to subcutaneous injections of either darbepoetin (dose adjusted to baseline Hb) or placebo every two weeks until they reached normal Hb levels (≥13 g/dL). Injections were given monthly thereafter to maintain Hb in the normal range. Despite a quick and sustained increase in Hb in the treatment group, darbepoetin had no effect on the primary outcome of death or hospitalisation with HF. There was a significantly higher rate of embolic and thrombotic events with darbepoetin compared to placebo (13.5% vs. 10%, p=0.009), which may have offset any potential benefit from darbepoetin.124
Similar neutral results for CV outcomes and increased risk of thrombotic events have been observed with ESAs in other patient populations.125 Consequently, ESAs are not used to treat anaemia in patients with HF. However, they continue to be used to treat anaemia in patients with CKD and the co-diagnosis of HF is not a contraindication.126
The FERRIC-HF (Effect of Intravenous Ferrous Sucrose on Exercise Capacity in Chronic Heart Failure), EFFECT-HF (Effect of Ferric Carboxymaltose on Exercise Capacity in Patients with Chronic Heart Failure and Iron Deficiency), FAIR-HF (Ferric Carboxymaltose in Patients with Heart Failure and Iron Deficiency), and CONFIRM-HF (Ferric Carboxymaltose Evaluation on Performance in Patients with Iron Deficiency in Combination with Chronic Heart Failure) trials investigated the safety and efficacy of IV iron in patients with HF and iron deficiency (ferritin <100 μg/L or <300 μg/L and transferrin saturation <20%).122,127–129
In the FERRIC-HF study, 35 patients (average age, 64; LVEF, ≤45%; NYHA II–III) were randomised in a 2:1 ratio to either placebo or IV iron, which was given weekly for four weeks and then at four-weekly intervals for 16 weeks.122 Despite a significant increase in ferritin and transferrin saturation in the iron group, there was no difference in the primary end point of change in peak exercise VO2 from baseline to 18 weeks compared to placebo.122
In the EFFECT-HF trial, 174 patients (average age, 63; average LVEF, 33%; median NT-proBNP, 1,576 ng/L in the treatment group) were randomised to either standard of care (no placebo) or IV iron, which was given to all patients at baseline and then at six-weekly intervals, depending on serum Hb.127 The primary end point was the difference in peak VO2 measured at baseline and 24 weeks. Peak VO2 decreased in the control group but was stable in the IV iron group and so, there was a small, but statistically significant difference in peak VO2 (+1.0 ml/kg/min; p=0.02) at the end of the study.127
In the FAIR-HF trial, 459 patients (average age, 67 years; average LVEF, 31%; approximately 80% of whom were NYHA class III) were randomised in a 2:1 ratio to either IV iron or placebo.128 Iron was given weekly until iron stores were normal and then every four weeks for a total of 24 weeks. The primary end point was a change in patient-reported symptoms or NYHA class. Half the patients in the iron group reported that their symptoms were ‘moderately’ or ‘much’ improved compared to 28% of patients in the placebo group (p<0.001). In a pre-planned subgroup analysis, IV iron was as effective for patients without anaemia (Hb ≥12 g/dL) as it was for patients with low Hb.130
Caution is required when considering IV iron for patients with HF. For example, in FAIR-HF, the proportion of patients in the placebo group who reported any improvement in symptoms (either ‘a little improved’, ‘moderately improved’ or ‘much improved’) was 53% compared to 74% in the IV iron group.128 This improvement was despite significant differences in ferritin (312 μg/L vs. 74 μg/L; p<0.001) and transferrin saturation (29% vs. 19%; p<0.001) between the groups after 24 weeks of treatment.127
In CONFIRM-HF, 301 patients (average age, 69 years; average LVEF, 37%; NYHA class II or III; average NT-proBNP, 2,511 pg/ml in the iron group) were randomised to either IV iron or placebo.129 Iron stores were replaced in an initial six-week treatment phase followed by further iron infusions every 12 weeks, only if the patient was still iron deficient. The primary outcome was change in 6MWT distance after 24 weeks of treatment; secondary outcomes included change in NYHA class, patient-reported symptoms and quality-of-life (QoL) scores. Follow up continued to one year.129
6MWT distance increased on average by 18 metres in the iron group and reduced by 16 metres in the placebo group, giving a difference in the change in 6MWT distance of 33 metres (p=0.002).129 Secondary end points were significantly improved in the treatment group compared to placebo. As with the results of FAIR-HF, IV iron was beneficial, regardless of whether or not the patients were anaemic.129
Although the trial was not powered to detect a difference, the CONFIRM-HF study was the first to suggest possible outcome benefit with IV iron; post-hoc analysis found that treatment with IV iron was associated with a lower risk of all-cause mortality or HF hospitalisation compared with placebo (HR 0.53; p=0.03).129
A subsequent individual patient data meta-analysis of 844 patients from four RCTs of IV iron (the majority from FAIR-HF and CONFIRM-HF) found that treatment with IV iron was associated with a lower rate of recurrent hospitalisation with HF (risk ratio [RR] 0.41; p=0.003) and lower rate of hospitalisation with HF or all-cause mortality (RR 0.54; p=0.011).130
More recently, two trials have suggested that there might be a benefit of IV iron on HF hospitalisations, but no effect on mortality. In the AFFIRM-AHF trial (Association Between Hemoglobin Levels and Efficacy of Intravenous Ferric Carboxymaltose in Patients with Acute Heart Failure and Iron Deficiency), 1,108 patients recruited soon after recovery from an episode of decompensated HF (average age, 71 years; 44% female; 50% NYHA class III; average LVEF, 33%; median NT-proBNP, 4,743 ng/L) were randomised to receive either IV ferric carboxymaltose or placebo.131 The primary end point was total HF hospitalisations or CV death after 12 months. There were fewer total HF hospitalisations in the IV iron group compared to placebo (RR 0.74; 95% CI, 0.58–0.94; p=0.035), but there was no impact on CV mortality (HR 0.96; 95% CI, 0.70–1.32; p=0.81) and the study was neutral for the primary end point (RR 0.79; 95% CI, 0.62–1.01; p=0.06).131
In the IRONMAN trial (Intravenous Ferric Derisomaltose in Patients with Heart Failure and Iron Deficiency in the UK), 1,137 patients with HF (average age 73 years; 25% female; 58% NYHA class II; median LVEF, 32%) were randomised to receive IV ferric derisomaltose or standard care.132 The trial was open label with masked adjudication of end points. The primary end point was recurrent hospital admissions for HF and CV death. After a median follow up of 2.7 years, IV iron was associated with a non-significant 5% absolute risk reduction (18% relative risk reduction) in the primary end point (p=0.07). In a COVID-19 (coronavirus disease 2019) sensitivity analysis during which all end points recorded after the onset of the COVID-19 pandemic were discarded, IV iron was associated with an 18% relative risk reduction in the primary end point (p=0.047). The benefit was driven by a reduction in HF hospitalisation; the rates of CV and all-cause mortality were not affected by IV iron.132
However, in the largest trial of IV iron in patients with HFrEF and iron deficiency by the ESC guideline definition – the HEART FID trial (Ferric Carboxymaltose in Heart Failure with Iron Deficiency) (n=1,532) – there was no difference in the rate of HF hospitalisation or mortality between the two treatment groups (mortality 8.6% vs. 10.3%; total HF hospitalisations 297 vs. 332 for IV iron and placebo, respectively; p=non-significant for both).133 It is likely that if IV iron has any effect on morbidity or mortality, the effect is small.
In IRONMAN, although patients were not blinded to treatment, QoL data was collected.132 IV iron was associated with a significant improvement in the Minnesota Living with Heart Failure symptom questionnaire score at four months after the first iron infusion (3.3 point difference; p=0.05) but by 20 months, the difference had disappeared.132 The IRONMAN investigators also assessed 6MWT distance in a subset of patients (n=193) at four and 20 months. In patients receiving standard of care, there was no change between the two time points (288 m at four months vs. 289 m at 20 months) but in patients receiving IV iron, the 6MWT distance fell by 33 metres (286 m at four months vs. 253 m at 20 months) with a p-value similar to that of the primary end point (p=0.068).132 IV iron clearly has a placebo effect, how much this contributes to the symptomatic benefit associated with IV iron is unknown.
Peak VO2 is the gold-standard measure of exercise capacity in patients with HF but was unchanged after 16 weeks of treatment with oral iron in the IRONOUT HF study (Iron Repletion Effects on Oxygen Uptake in Heart Failure) with only small and (likely) clinically insignificant changes reported in the FERRIC-HF (Effect of Intravenous Ferrous Sucrose on Exercise Capacity in Chronic Heart Failure) and EFFECT-HF (Effect of Ferric Carboxymaltose on Exercise Capacity in Patients with Chronic Heart Failure and Iron Deficiency) trials.122,127,134 It is not defined as a primary or secondary outcome for the ongoing studies of IV iron and so whether or not IV iron improves exercise capacity in patients with HF as measured by peak VO2 will remain unsettled.
There is one more ongoing trial of IV iron in patients with HF and iron deficiency yet to complete: FAIR-HF 2 (Intravenous Iron in Patients with Systolic Heart Failure and Iron Deficiency to Improve Morbidity and Mortality 2).135
Regular IV iron infusions are expensive and logistically challenging. By comparison, iron tablets are cheap and widely available, yet were not investigated for the treatment of iron deficiency in patients with HF until 2016.
In the IRONOUT study, 225 patients (ferritin <100 μg/L or <300 μg/L and transferrin saturation <20%; average age, 63 years; NYHA II–III; median LVEF, 25%; median NT-proBNP, 1,111 pg/mL; median Hb, 12.6 g/dL) were randomised to either iron polysaccharide 150 mg twice daily or placebo for 16 weeks.134 The primary outcome was the change in peak exercise VO2. Secondary outcome measures included other exercise variables, 6MWT distance, NT-proBNP and QoL measures. The study was neutral for all primary and secondary outcomes.134
While transferrin saturation significantly improved (p=0.003), the median increment from baseline at 16 weeks for patients treated with oral iron versus placebo was very small (3%) and most patients in the oral iron group remained iron deficient (by the investigators’ definition) after 16 weeks of treatment (median ferritin, 95 ng/L).134 Such small improvements are unlikely to translate into a clinically detectable difference, suggesting that the duration, dose, route of administration of iron, or all three were insufficient to improve iron stores in patients with HF.134
Summary
Managing a patient with HF is as much about managing the complications of the disease and its treatment as it is the condition itself. Electrolyte abnormalities, hypotension, and renal dysfunction are common and may impair the efficacy of treatment, worsen quality of life, and lead to adverse outcomes. There are few strategies to combat each complication and more research is needed but that does not mean solutions cannot be found in some cases. Finally, although iron deficiency is common and treatment with IV iron common practice, there is no robust data demonstrating meaningful improvements in quality of life, morbidity, or mortality despite many thousands of patients randomised in multiple clinical trials.
close window and return to take test
References
- Ahmed M, Ekundayo O, Mujib M et al. Mild hyperkalemia and outcomes in chronic heart failure: a propensity matched study. Int J Cardiol 2010;144:383–8. https://doi.org/10.1016/j.ijcard.2009.04.041 [Epub ahead of print]
- Gheorghiade M, Rossi J, Cotts W et al. Characterization and prognostic value of persistent hyponatremia in patients with severe heart failure in the escape trial. Arch Intern Med 2007;167:1998–2005. https://doi.org/10.1001/archinte.167.18.1998
- Cooper L, Mentz R, Gallup D et al. Serum bicarbonate in acute heart failure: relationship to treatment strategies and clinical outcomes. J Card Fail 2016;22:738–42. https://doi.org/10.1016/j.cardfail.2016.01.007 [Epub ahead of print]
- Krogager ML, Eggers-Kaas L, Aasbjerg K et al. Short-term mortality risk of serum potassium levels in acute heart failure following myocardial infarction. Eur Heart J Cardiovasc Pharmacother 2015;1:245–51. https://doi.org/10.1093/ehjcvp/pvv026 [Epub ahead of print]
- Hoss S, Elizur Y, Luria D, Keren A, Lotan C, Gotsman I. Serum potassium levels and outcome in patients with chronic heart failure. Am J Cardiol 2016;118:1868–74. https://doi.org/10.1016/j.amjcard.2016.08.078 [Epub ahead of print]
- Ter Maaten JM, Damman K, Hanberg JS et al. Hypochloremia, diuretic resistance, and outcome in patients with acute heart failure. Circ Heart Fail 2016;9:e003109. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003109
- Cuthbert JJ, Pellicori P, Rigby A et al. Low serum chloride in patients with chronic heart failure: clinical associations and prognostic significance. Eur J Heart Fail 2018;20:1426–35. https://doi.org/10.1002/ejhf.1247 [Epub ahead of print]
- Urso C, Brucculeri S, Caimi G. Acid-base and electrolyte abnormalities in heart failure: pathophysiology and implications. Heart Fail Rev 2015;20:493–503. https://doi.org/10.1007/s10741-015-9482-y
- Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 8: Advanced challenges in resuscitation: section 1: life-threatening electrolyte abnormalities. The American Heart Association in collaboration with the International Liaison Committee on Resuscitation. Circulation 2000;102(8 Suppl):I217–22.
- Nicholas R, Banner NR, Bonser RS et al. UK guidelines for referral and assessment of adults for heart transplantation. Heart 2011;97:1520–7. https://doi.org/10.1136/heartjnl-2011-300048
- Verbrugge FH, Steels P, Grieten L, Nijst P, Tang WH, Mullens W. Hyponatremia in acute decompensated heart failure: depletion versus dilution. J Am Coll Cardiol 2015;65:480–92. https://doi.org/10.1016/j.jacc.2014.12.010
- Clark AL, Gardner RS, McDonagh TA (eds.). Oxford Textbook of Heart Failure (2nd edn.). Oxford: Oxford University Press, 2022.
- Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. Am J Med 2006;119(Suppl 1):S47–53. https://doi.org/10.1016/j.amjmed.2006.05.007
- Schrier RW, Berl T. Nonosmolar factors affecting renal water excretion (first of two parts). N Engl J Med 1975;292:81–8. https://doi.org/10.1056/NEJM197501092920207
- Felker GM, O’Connor CM, Braunwald E; Heart Failure Clinical Research Network Investigators. Loop diuretics in acute decompensated heart failure: A necessary evil? Circ Heart Fail 2009;2:56–62. https://doi.org/10.1161/CIRCHEARTFAILURE.108.821785
- Verbrugge FH, Dupont M, Steels P et al. The kidney in congestive heart failure: ‘are natriuresis, sodium, and diuretics really the good, the bad and the ugly?’ Eur J Heart Fail 2014:16;133–42. https://doi.org/10.1002/ejhf.35 [Epub ahead of print]
- National Institute for Cardiovascular Outcomes Research. The National Heart Failure Audit. Published 2023 Summary Report (2021/22 data). 2023. Available from: https://www.nicor.org.uk/wp-content/uploads/2023/10/10633-NICOR-Annual-Summary_Reports_NHFA_v5.AC_.pdf (accessed 11 January 2024)
- Rusinaru D, Tribouilloy C, Berry C et al; MAGGIC Investigators. Relationship of serum sodium concentration to mortality in a wide spectrum of heart failure patients with preserved and with reduced ejection fraction: an individual patient data meta-analysis: Meta-Analysis Global Group in Chronic heart failure (MAGGIC). Eur J Heart Fail 2012;14:1139–46. https://doi.org/10.1093/eurjhf/hfs099 [Epub ahead of print]
- Klein L, O’Connor CM, Leimberger JD et al; OPTIME-CHF Investigators. Lower serum sodium is associated with increased short-term mortality in hospitalized patients with worsening heart failure: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) study. Circulation 2005;111:2454–60. https://doi.org/10.1161/01.CIR.0000165065.82609.3D [Epub ahead of print]
- De Vecchis R, Di Maio M, Di Biase G, Ariano C. Effects of hyponatremia normalization on the short-term mortality and rehospitalizations in patients with recent acute decompensated heart failure: a retrospective study. J Clin Med 2016;5:92. https://doi.org/10.3390/jcm5100092
- McDonagh TA, Metra M, Adamo M et al; ESC Scientific Document Group. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021;42:3599–726. https://doi.org/10.1093/eurheartj/ehab368. Erratum in: Eur Heart J. 2021 Oct 14.
- Aliti GB, Rabelo ER, Clausell N, Rohde LE, Biolo A, Beck-da-Silva L. Aggressive fluid and sodium restriction in acute decompensated heart failure: a randomized clinical trial. JAMA Intern Med 2013;173:1058–64. https://doi.org/10.1001/jamainternmed.2013.552
- Paterna S, Gaspare P, Fasullo S, Sarullo FM, Di Pasquale P. Normal-sodium diet compared with low-sodium diet in compensated congestive heart failure: is sodium an old enemy or a new friend? Clin Sci (London) 2008;114:221–30. https://doi.org/10.1042/CS20070193
- Paterna S, Di Pasquale P, Parrinello G et al. Changes in brain natriuretic peptide levels and bioelectrical impedance measurements after treatment with high-dose furosemide and hypertonic saline solution versus high-dose furosemide alone in refractory congestive heart failure: a double-blind study. J Am Coll Cardiol 2005;45:1997–2003. https://doi.org/10.1016/j.jacc.2005.01.059
- Licata G, Di Pasquale P, Parrinello G et al. Effects of high-dose furosemide and small-volume hypertonic saline solution infusion in comparison with a high dose of furosemide as bolus in refractory congestive heart failure: long-term effects. Am Heart J 2003;145:459–66. https://doi.org/10.1067/mhj.2003.166
- Okuhara Y, Hirotani S, Naito Y et al. Intravenous salt supplementation with low-dose furosemide for treatment of acute decompensated heart failure. J Card Fail 2014;20:295–301. https://doi.org/10.1016/j.cardfail.2014.01.012 [Epub ahead of print]
- Gheorghiade M, Konstam M, Grinfeld L et al; EVEREST Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST clinical status trials. JAMA 2007;297:1332–43. https://doi.org/10.1001/jama.297.12.1332 [Epub ahead of print]
- Gheorghiade M, Gottlieb S, Udelson J et al; Tolvaptan Investigators. Vasopressin V(2) receptor blockade with tolvaptan versus fluid restriction in the treatment of hyponatremia. Am J Cardiol 2006;97:1064–7. https://doi.org/10.1016/j.amjcard.2005.10.050 [Epub ahead of print]
- Konstam MA, Gheorghiade M, Burnett JC Jr et al; EVEREST Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007;297:1319–31. https://doi.org/10.1001/jama.297.12.1319 [Epub ahead of print]
- Felker GM, Mentz RJ, Cole RT et al. Efficacy and safety of tolvaptan in patients hospitalized with acute heart failure (TACTICS-HF). J Am Coll Cardiol 2017;69:1399–406. https://doi.org/10.1016/j.jacc.2016.09.004 [Epub ahead of print]
- Konstam MA, Kiernan M, Chandler A et al; SECRET of CHF Investigators, Coordinators, and Committee Members. Short-term effects of tolvaptan in patients with acute heart failure and volume overload (SECRET of CHF). J Am Coll Cardiol 2017;69:1409–19. https://doi.org/10.1016/j.jacc.2016.12.035
- Hauptman PJ, Burnett J, Gheorghiade M et al; EVEREST Investigators. Clinical course of patients with hyponatremia and decompensated systolic heart failure and the effect of vasopressin receptor antagonism with tolvaptan. J Card Fail 2013;19:390–7. https://doi.org/10.1016/j.cardfail.2013.04.001 [Epub ahead of print]
- Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol 1990;65:4E–9E. https://doi.org/10.1016/0002-9149(90)90244-u
- Weiss JN, Qu Z, Shivkumar K. Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol 2017;10:e004667. https://doi.org/10.1161/CIRCEP.116.004667
- Pezhouman A, Singh N, Song Z et al. Molecular basis of hypokalemia-induced ventricular fibrillation. Circulation 2015;132:1528–37. https://doi.org/10.1161/CIRCULATIONAHA.115.016217 [Epub ahead of print]
- Jahnen-Dechent W, Ketteler M. Magnesium basics. Clin Kidney J 2012;5(Suppl 1):i3–i14. https://doi.org/10.1093/ndtplus/sfr163
- Palmer BF. Regulation of potassium homeostasis. Clin J Am Soc Nephrol 2015;10:1050–60. https://doi.org/10.2215/CJN.08580813
- Palmer BF. Managing hyperkalemia caused by inhibitors of the renin-angiotensin-aldosterone system. N Engl J Med 2004;351:585–92. https://doi.org/10.1056/NEJMra035279
- Rabelink TJ, Koomans HA, Hené RJ et al. Early and late adjustment to potassium loading in humans. Kidney Int 1990;38:942–7. https://doi.org/10.1038/ki.1990.295
- Zannad F. Aldosterone and heart failure. Eur Heart J 1995;16(Suppl N):98–102. https://doi.org/10.1093/eurheartj/16.suppl_n.98
- Maggioni AP, Anker SD, Dahlstrom U et al. Are hospitalized or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12,440 patients of the ESC Heart Failure Long-Term Registry. Eur J Heart Fail 2013;15:1173−84. https://doi.org/10.1093/eurjhf/hft134 [Epub ahead of print]
- Weir MR, Rolfe M. Potassium homeostasis and renin-angiotensin-aldosterone system inhibitors. Clin J Am Soc Nephrol 2010;5:531−48. https://doi.org/10.2215/CJN.07821109 [Epub ahead of print]
- Reardon LC, Macpherson DS. Hyperkalemia in outpatients using angiotensin-converting enzyme inhibitors. How much should we worry? Arch Intern Med 1998;158:26–32. https://doi.org/10.1001/archinte.158.1.26
- Yusuf S, Pitt B, Davis CE, Hood WB, Cohn JN; SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991;325:293–302. https://doi.org/10.1056/NEJM199108013250501
- McMurray JJV, Östergren J, Swedberg K et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet 2003;362:767–71.
- Pitt B, Zannad F, Remme WJ et al; Randomized Aldactone Evaluation Study (RALES) Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999;341:709–17.
- Zannad F, McMurray JJV, Krum H et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011;364:11−21. https://doi.org/10.1056/NEJMoa1009492 [Epub ahead of print]
- McMurray JJV, Packer M, Desai AS et al; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993–1004. https://doi.org/10.1056/NEJMoa1409077 [Epub ahead of print]
- Vukadinović D, Lavall D, Vukadinović AN, Pitt B, Wagenpfeil S, Böhm M. True rate of mineralocorticoid receptor antagonists-related hyperkalemia in placebo-controlled trials: A meta-analysis. Am Heart J 2017;188:99−108. https://doi.org/10.1016/j.ahj.2017.03.011 [Epub ahead of print]
- Vardeny O, Claggett B, Anand I et al; RALES Investigators. Incidence, predictors, and outcomes related to hypo- and hyperkalemia in patients with severe heart failure treated with a mineralocorticoid receptor antagonist. Circ Heart Fail 2014;7:573−9. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001104 [Epub ahead of print]
- Rossignol P, Dobre D, McMurray JJV et al; EMPHASIS-HF Study Group. Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo in addition to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail 2014;7:51−8. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000792 [Epub ahead of print]
- National Institute for Health and Care Excellence. Chronic heart failure in adults: diagnosis and management [NG106]. London: NICE, 2018. Available from: https://www.nice.org.uk/guidance/ng106/chapter/Recommendations#monitoring-treatment-for-all-types-of-heart-failure (accessed 29 January 2024)
- National Institute for Health and Care Excellence. Patiromer for treating hyperkalaemia. Technology appraisal guidance [TA623]. London: NICE, 2020. Available from: https://www.nice.org.uk/guidance/ta623/resources/patiromer-for-treating-hyperkalaemia-pdf-82609015577029 (accessed 30 October 2023)
- National Institute for Health and Care Excellence. Sodium zirconium cyclosilicate for treating hyperkalaemia. Technology appraisal guidance [TA599]. London: NICE, 2022. Available from: https://www.nice.org.uk/guidance/ta599/chapter/1-Recommendations (accessed 30 October 2023)
- Harel Z, Harel S, Shah PS, Wald R, Perl J, Bell CM. Gastrointestinal adverse events with sodium polystyrene sulfonate (Kayexalate) use: a systematic review. Am J Med 2013;126:264.e9–24. https://doi.org/10.1016/j.amjmed.2012.08.016 [Epub ahead of print]
- Domanski M, Norman J, Pitt B, Haigney M, Hanlon S, Peyster E. Diuretic use, progressive heart failure, and death in patients in the Studies Of Left Ventricular Dysfunction (SOLVD). J Am Coll Cardiol 2003;42:705–8. https://doi.org/10.1016/s0735-1097(03)00765-4
- Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ; PEARL-HF Investigators. Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double-blind, placebo-controlled study in patients with chronic heart failure (the PEARL-HF) trial. Eur Heart J 2011;32:820–8. https://doi.org/10.1093/eurheartj/ehq502 [Epub ahead of print]
- Pitt B, Bushinsky DA, Kitzman DW, Ruschitzka F et al; Patiromer-204 Investigators. Evaluation of an individualized dose titration regimen of patiromer to prevent hyperkalaemia in patients with heart failure and chronic kidney disease. ESC Heart Fail 2018;5:257–66. https://doi.org/10.1002/ehf2.12265 [Epub ahead of print]
- Butler J, Anker SD, Lund LH et al. Patiromer for the management of hyperkalemia in heart failure with reduced ejection fraction: the DIAMOND trial. Eur Heart J 2022;43:4362–73. https://doi.org/10.1093/eurheartj/ehac401
- Kosiborod M, Rasmussen HS, Lavin P et al. Effect of sodium zirconium cyclosilicate on potassium lowering for 28 days among outpatients with hyperkalemia: the HARMONIZE randomized clinical trial. JAMA 2014;312:2223–33. https://doi.org/10.1001/jama.2014.15688
- Anker SD, Kosiborod M, Zannad F et al. Maintenance of serum potassium with sodium zirconium cyclosilicate (ZS-9) in heart failure patients: results from a phase 3 randomized, double-blind, placebo-controlled trial. Eur J Heart Fail 2015;17:1050–6. https://doi.org/10.1002/ejhf.300 [Epub ahead of print]
- Kosiborod M, Peacock WF, Packham DK. Sodium zirconium cyclosilicate for urgent therapy of severe hyperkalemia. N Engl J Med 2015;372:1577–8. https://doi.org/10.1056/NEJMc1500353
- Zannad F, Hsu BG, Maeda Y et al. Efficacy and safety of sodium zirconium cyclosilicate for hyperkalaemia: the randomized, placebo-controlled HARMONIZE-global study. ESC Heart Fail 2020;7:54–64. https://doi.org/10.1002/ehf2.12561 [Epub ahead of print]
- Crespo-Leiro MG, Anker SD, Maggioni AP et al; Heart Failure Association (HFA) of the European Society of Cardiology (ESC). European Society of Cardiology Heart Failure Long-Term Registry (ESC-HF-LT): 1-year follow-up outcomes and differences across regions. Eur J Heart Fail 2016;18:613–25. https://doi.org/10.1002/ejhf.566
- Cleland JGF, Swedberg K, Follath F et al; Study Group on Diagnosis of the Working Group on Heart Failure of the European Society of Cardiology. The EuroHeart Failure survey programme – a survey on the quality of care among patients with heart failure in Europe. Part 1: patient characteristics and diagnosis. Eur Heart J 2003;24:442–63. https://doi.org/10.1016/s0195-668x(02)00823-0
- de Silva R, Rigby AS, Witte KKA et al. Anemia, renal dysfunction, and their interaction in patients with chronic heart failure. Am J Cardiol 2006;98:391–8. https://doi.org/10.1016/j.amjcard.2006.01.107 [Epub ahead of print]
- Hillege HL, Nitsch D, Pfeffer MA et al; CHARM Investigators. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation 2006;113:671–8. https://doi.org/10.1161/CIRCULATIONAHA.105.580506
- Hillege HL, Girbes AR, de Kam PJ et al. Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 2000;102:203–10. https://doi.org/10.1161/01.cir.102.2.203
- Farré N, Vela E, Clèries M et al. Real world heart failure epidemiology and outcome: A population-based analysis of 88,195 patients. PLoS One 2017;12:e0172745. https://doi.org/10.1371/journal.pone.0172745
- de Silva R, Nikitin NP, Witte KKA et al. Incidence of renal dysfunction over 6 months in patients with chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J 2006;27:569–81. https://doi.org/10.1093/eurheartj/ehi696 [Epub ahead of print]
- Abuelo JG. Normotensive ischemic acute renal failure. N Engl J Med 2007;357:797–805. https://doi.org/10.1056/NEJMra064398
- Liu YL, Prowle J, Licari E, Uchino S, Bellomo R. Changes in blood pressure before the development of nosocomial acute kidney injury. Nephrol Dial Transplant 2009;24:504–11. https://doi.org/10.1093/ndt/gfn490 [Epub ahead of print]
- Firth JF, Raine AE, Ledingham JG. Raised venous pressure: a direct cause of renal sodium retention in oedema? Lancet 1988;1:1033–5.
- Mullens W, Abrahams Z, Skouri HN et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol 2008;51:300–6. https://doi.org/10.1016/j.jacc.2007.09.043
- de Silva R, Loh H, Rigby AS et al. Epidemiology, associated factors, and prognostic outcomes of renal artery stenosis in chronic heart failure assessed by magnetic resonance angiography. Am J Cardiol 2007;100:273–9. https://doi.org/10.1016/j.amjcard.2007.02.098 [Epub ahead of print]
- Gupta R, Syed M, Ashcherkin N, Chen K, Vaidya PP, Cooper CJ. Renal artery stenosis and congestive heart failure: what do we really know? Curr Cardiol Rep 2019;21:74. https://doi.org/10.1007/s11886-019-1169-x
- Devoy MA, Tomson CR, Edmunds ME, Feehally J, Walls J. Deterioration in renal function associated with angiotensin converting enzyme inhibitor therapy is not always reversible. J Intern Med 1992;232:493–8. https://doi.org/10.1111/j.1365-2796.1992.tb00622.x
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med 1987;316:1429–35. https://doi.org/10.1056/NEJM198706043162301
- Ljungman S, Kjekshus J, Swedberg K. Renal function in severe congestive heart failure during treatment with enalapril (the Cooperative North Scandinavian Enalapril Survival Study [CONSENSUS] Trial). Am J Cardiol 1992;70:479–87. https://doi.org/10.1016/0002-9149(92)91194-9
- Yusuf S, Pitt B, Davis CE, Hood WB Jr, Cohn JN; SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med 1992;327:685–91. https://doi.org/10.1056/NEJM199209033271003
- Testani JM, Kimmel SE, Dries DL, Coca SG. Prognostic importance of early worsening renal function after initiation of angiotensin-converting enzyme inhibitor therapy in patients with cardiac dysfunction. Circ Heart Fail 2011;4:685–91. https://doi.org/10.1161/CIRCHEARTFAILURE.111.963256 [Epub ahead of print]
- Vardeny O, Wu DH, Desai A et al; RALES Investigators. Influence of baseline and worsening renal function on efficacy of spironolactone in patients with severe heart failure: insights from RALES (Randomized Aldactone Evaluation Study). J Am Coll Cardiol 2012;60:2082–9. https://doi.org/10.1016/j.jacc.2012.07.048 [Epub ahead of print]
- Clark H, Krum H, Hopper I. Worsening renal function during renin-angiotensin-aldosterone system inhibitor initiation and long-term outcomes in patients with left ventricular systolic dysfunction. Eur J Heart Fail 2014;16:41–8. https://doi.org/10.1002/ejhf.13 [Epub ahead of print]
- Damman K, Valente MA, Voors AA et al. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J 2014;35:455–69. https://doi.org/10.1093/eurheartj/eht386 [Epub ahead of print]
- Brisco MA, Zile MR, Hanberg JS et al. Relevance of changes in serum creatinine during a heart failure trial of decongestive strategies: insights from the DOSE Trial. J Card Fail 2016;22:753–60. https://doi.org/10.1016/j.cardfail.2016.06.423 [Epub ahead of print]
- Metra M, Davison B, Bettari L et al. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ Heart Fail 2012;5:54–62. https://doi.org/10.1161/CIRCHEARTFAILURE.111.963413 [Epub ahead of print]
- National Institute of Health and Care Excellence. Chronic kidney disease in adults: assessment and management. Clinical guideline [CG182]. London: NICE, 2014. Available from: https://www.nice.org.uk/guidance/CG182 (accessed 30 October 2023)
- Clark AL, Kalra PR, Petrie MC, Mark PB, Tomlinson LA, Tomson CR. Change in renal function associated with drug treatment in heart failure: national guidance. Heart 2019;105:904–10. https://doi.org/10.1136/heartjnl-2018-314158
- Pellicori P, Kalra PR, Clark AL, Friday JM, Cleland JGF. Chronic kidney disease (CKD) and CKD-ism in heart failure – what a mess! Eur J Heart Fail 2022;24:2196–8. https://doi.org/10.1002/ejhf.2696 [Epub ahead of print]
- O’Meara E, Rouleau JL, White M et al. Heart failure with anemia: novel findings on the roles of renal disease, interleukins, and specific left ventricular remodeling processes. Circ Heart Fail 2014;7:773–81. https://doi.org/10.1161/CIRCHEARTFAILURE.114.001100 [Epub ahead of print]
- Cleland JG, Zhang J, Pellicori P et al. Prevalence and outcomes of anemia and hematinic deficiencies in patients with chronic heart failure. JAMA Cardiol 2016;1:539–47. https://doi.org/10.1001/jamacardio.2016.1161
- Jankowska EA, von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. Eur Heart J 2013;34:816–29. https://doi.org/10.1093/eurheartj/ehs224 [Epub ahead of print]
- Groenveld HF, Januzzi JL, Damman K et al. Anemia and mortality in heart failure patients a systematic review and meta-analysis. J Am Coll Cardiol 2008;52:818–27. https://doi.org/10.1016/j.jacc.2008.04.061
- Opasich C, Cazzola M, Scelsi L et al. Blunted erythropoietin production and defective iron supply for erythropoiesis as major causes of anaemia in patients with chronic heart failure. Eur Heart J 2005;26:2232–7. https://doi.org/10.1093/eurheartj/ehi388 [Epub ahead of print]
- Nanas JN, Matsouka C, Karageorgopoulos D et al. Etiology of anemia in patients with advanced heart failure. J Am Coll Cardiol 2006;48:2485–9. https://doi.org/10.1016/j.jacc.2006.08.034 [Epub ahead of print]
- Montalescot G, Sechtem U, Achenbach S et al. 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J 2013;34:2949–3003. https://doi.org/10.1093/eurheartj/eht296 [Epub ahead of print]
- Komajda M, Follath F, Swedberg K et al; Study Group on Diagnosis of the Working Group on Heart Failure of the European Society of Cardiology. The EuroHeart Failure Survey programme — a survey on the quality of care among patients with heart failure in Europe. Part 2: treatment. Eur Heart J 2003;24:464–74. https://doi.org/10.1016/s0195-668x(02)00700-5
- Goddard AF, James MW, McIntyre AS, Scott BB; British Society of Gastroenterology. Guidelines for the management of iron deficiency anaemia. Gut 2011;60:1309–16. https://doi.org/10.1136/gut.2010.228874 [Epub ahead of print]
- Dunn LL, Rahmanto YS, Richardson DR. Iron uptake and metabolism in the new millennium. Trends Cell Biol 2007;17:93–100. https://doi.org/10.1016/j.tcb.2006.12.003 [Epub ahead of print]
- Beard JL. Iron biology in immune function, muscle metabolism and neuronal functioning. J Nutr 2001;131:568S–80S. https://doi.org/10.1093/jn/131.2.568S
- Jankowska EA, Rozentryt P, Witkowska A et al. Iron deficiency: an ominous sign in patients with systolic chronic heart failure. Eur Heart J 2010;31:1872–80. https://doi.org/10.1093/eurheartj/ehq158 [Epub ahead of print]
- Klip IT, Comin‐Colet J, Voors AA et al. Iron deficiency in chronic heart failure: an international pooled analysis. Am Heart J 2013;165:575–82. https://doi.org/10.1016/j.ahj.2013.01.017 [Epub ahead of print]
- Isakov E, Froom P, Henig C, Barak M. Anemia and estimated glomerular filtration rates. Ann Clin Lab Sci 2014;44:419–24. http://www.annclinlabsci.org/content/44/4/419.long
- Ishani A, Weinhandl E, Zhao Z et al. Angiotensin-converting enzyme inhibitors as a risk factor for the development of anemia, and the impact of incident anemia on mortality in patients with left ventricular dysfunction. J Am Coll Cardiol 2005;45:391–9. https://doi.org/10.1016/j.jacc.2004.10.038
- Mrug M, Stopka T, Julian BA, Prchal JF, Prchal JT. Angiotensin II stimulates proliferation of normal early erythroid progenitors. J Clin Invest 1997:100;2310–4. https://doi.org/10.1172/JCI119769
- Van der Meer P, Lipsic E, Westenbrink BD et al. Levels of hematopoiesis inhibitor N-acetyl-seryl-aspartyl-lysyl-proline partially explain the occurrence of anemia in heart failure. Circulation 2005;112:1743–7. https://doi.org/10.1161/CIRCULATIONAHA.105.549121
- Fildes JE, Shaw SM, Yonan N, Williams SG. The immune system and chronic heart failure: is the heart in control? J Am Coll Cardiol 2009;53:1013–20. https://doi.org/10.1016/j.jacc.2008.11.046
- Rauchhaus M, Doehner W, Francis DP et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation 2000;102:3060–7. https://doi.org/10.1161/01.cir.102.25.3060
- Ferrucci L, Guralnik JM, Woodman RC et al. Proinflammatory state and circulating erythropoietin in persons with and without anemia. Am J Med 2005;118:1288. https://doi.org/10.1016/j.amjmed.2005.06.039
- Deicher R, Horl WH. Hepcidin: a molecular link between inflammation and anaemia. Dial Transplant 2004;19:521–4. https://doi.org/10.1093/ndt/gfg560
- Androne AS, Katz SD, Lund L et al. Hemodilution is common in patients with advanced heart failure. Circulation 2003;107:226–9. https://doi.org/10.1161/01.cir.0000052623.16194.80
- Westenbrink BD, Visser FW, Voors AA et al. Anaemia in chronic heart failure is not only related to impaired renal perfusion and blunted erythropoietin production, but to fluid retention as well. Eur Heart J 2007;28:166–71. https://doi.org/10.1093/eurheartj/ehl419 [Epub ahead of print]
- Sze S, Pellicori P, Kazmi S et al. Prevalence and prognostic significance of malnutrition using 3 scoring systems among outpatients with heart failure: a comparison with body mass index. JACC Heart Fail 2018;6:476–86. https://doi.org/10.1016/j.jchf.2018.02.018 [Epub ahead of print]
- Sze S, Pellicori P, Zhang J, Clark AL. Malnutrition, congestion and mortality in ambulatory patients with heart failure. Heart 2019;105:297–306. https://doi.org/10.1136/heartjnl-2018-313312 [Epub ahead of print]
- World Health Organisation. The global prevalence of anaemia in 2011. 2015. Available from: http://apps.who.int/iris/bitstream/handle/10665/177094/9789241564960_eng.pdf;jsessionid=7F366C8F8D366296C40C469143631C65?sequence=1 (accessed 30 October 2023)
- Witte KKA, Desilva R, Chattopadhyay S, Ghosh J, Cleland JG, Clark AL. Are hematinic deficiencies the cause of anemia in chronic heart failure? Am Heart J 2004;147:924–30. https://doi.org/10.1016/j.ahj.2003.11.007
- van der Wal HH, Comin-Colet J, Klip IT et al. Vitamin B12 and folate deficiency in chronic heart failure. Heart 2015;101:302–10. https://doi.org/10.1136/heartjnl-2014-306022 [Epub ahead of print]
- Cromie N, Lee C, Struthers AD. Anaemia in chronic heart failure: what is its frequency in the UK and its underlying causes? Heart 2002;87:377–8. https://doi.org/10.1136/heart.87.4.377
- Yates JM, Logan EC, Stewart RM. Iron deficiency anaemia in general practice: clinical outcomes over three years and factors influencing diagnostic investigations. Postgrad Med J 2004;80:405–10. https://doi.org/10.1136/pgmj.2003.015677
- Lucas CA, Logan EC, Logan RF. Audit of the investigation and outcome of iron-deficiency anaemia in one health district. J R Coll Physicians Lond 1996;30:33–6.
- van Veldhuisen DJ, Dickstein K, Cohen-Solal A et al. Randomized, double-blind, placebo-controlled study to evaluate the effect of two dosing regimens of darbepoetin alfa in patients with heart failure and anaemia. Eur Heart J 2007;28:2208–16. https://doi.org/10.1093/eurheartj/ehm328 [Epub ahead of print]
- Okonko DO, Grzeslo A, Witkowski T et al. Effect of intravenous iron sucrose on exercise tolerance in anemic and nonanemic patients with symptomatic chronic heart failure and iron deficiency FERRIC-HF: a randomized, controlled, observer-blinded trial. J Am Coll Cardiol 2008;51:103–12. https://doi.org/10.1016/j.jacc.2007.09.036
- Silverberg DS, Wexler D, Sheps D et al. The effect of correction of mild anemia in severe, resistant congestive heart failure using subcutaneous erythropoietin and intravenous iron: a randomized controlled study. J Am Coll Cardiol 2001;37:1775–80. https://doi.org/10.1016/s0735-1097(01)01248-7
- Swedberg K, Young JB, Anand IS et al; RED-HF Investigators. Treatment of anemia with darbepoetin alfa in systolic heart failure. N Engl J Med 2013;368:1210–9. https://doi.org/10.1056/NEJMoa1214865 [Epub ahead of print]
- Singh AK, Szczech L, Tang KL et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med 2006;355:2085–98. https://doi.org/10.1056/NEJMoa065485
- KDIGO. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease 2012. Available at: http://kdigo.org/clinical_practice_guidelines/pdf/KDIGO-Anemia%20GL.pdf (accessed 31 October 2023)
- van Veldhuisen DJ, Ponikowski P, van der Meer P et al; EFFECT-HF Investigators. Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation 2017;136:1374–83. https://doi.org/10.1161/CIRCULATIONAHA.117.027497 [Epub ahead of print]
- Anker SD, Comin Colet J, Filippatos G et al; FAIR-HF Trial Investigators. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009;361:2436–48. https://doi.org/10.1056/NEJMoa0908355 [Epub ahead of print]
- Ponikowski P, van Veldhuisen DJ, Comin-Colet J et al; CONFIRM-HF Trial Investigators. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur Heart J 2015;36:657–68. https://doi.org/10.1093/eurheartj/ehu385 [Epub ahead of print]
- Anker SD, Kirwan BA, van Veldhuisen DJ et al. Effects of ferric carboxymaltose on hospitalisations and mortality rates in iron-deficient heart failure patients: an individual patient data meta-analysis. Eur J Heart Fail 2018;20:125–33. https://doi.org/10.1002/ejhf.823 [Epub ahead of print]
- Ponikowski P, Kirwan BA, Anker SD et al; AFFIRM-AHF Investigators. Ferric carboxymaltose for iron deficiency at discharge after acute heart failure: a multicentre, double-blind, randomised, controlled trial. Lancet 2020;396:1895–904. https://doi.org/10.1016/S0140-6736(20)32339-4
- Kalra PR, Cleland JGF, Petrie MC et al; IRONMAN Trial Investigators. Intravenous ferric derisomaltose in patients with heart failure and iron deficiency in the UK (IRONMAN): an investigator-initiated, prospective, randomised, open-label, blinded-end point trial. Lancet 2022;400:2199–209. https://doi.org/10.1016/S0140-6736(22)02083-9 [Epub ahead of print]
- Mentz RJ, Garg J, Rockhold FW et al. Ferric Carboxymaltose in Heart Failure with Iron Deficiency. N Engl J Med 2023;389(11):975-986. https://doi.org/10.1056/NEJMoa2304968
- Lewis GD, Malhotra R, Hernandez AF et al; NHLBI Heart Failure Clinical Research Network. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA 2017;317:1958–66. https://doi.org/10.1001/jama.2017.5427
- ClinicalTrials.gov. Intravenous iron in patients with systolic heart failure and iron deficiency to improve morbidity & mortality. Available at: https://classic.clinicaltrials.gov/ct2/show/NCT03036462?term=1.%09NCT03036462&draw=2&rank=1 (accessed 29 January 2024)

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.