Introduction
Table 1. Different types of dyslipidaemia
| Class of dyslipidaemia | Lipid pattern |
| Hypercholesterolaemia | ↑ total cholesterol ↑ LDL-C |
| Hypertriglyceridemia | ↑ triglyceride levels |
| Mixed dyslipidaemia | ↑ total cholesterol and ↑ triglyceride levels |
| Key: LDL-C = low-density lipoprotein cholesterol | |
Dyslipidaemias can be considered primary lipid disorders or secondary to an underlying disease or medication. They can be subcategorised further based on the pattern of lipoprotein disturbance observed upon lipid profiling (table 1), which is important when considering the underlying cause.
For the majority of people with dyslipidaemia, adequate control can be achieved with lifestyle measures and pharmacotherapy, of which statins are the mainstay of initial medical treatment.1,2 Clinicians must always consider the possibility of a familial cause, especially in patients with premature atherosclerosis as well as a strong family history of vascular and coronary heart disease (CHD).3 Abnormalities in serum lipoprotein concentrations are found in seven of out every 10 patients with premature CHD, with a familial disorder in more than half of these cases. Unfortunately, inherited lipid disorders may not be identified early and are commonly underdiagnosed in routine practice.
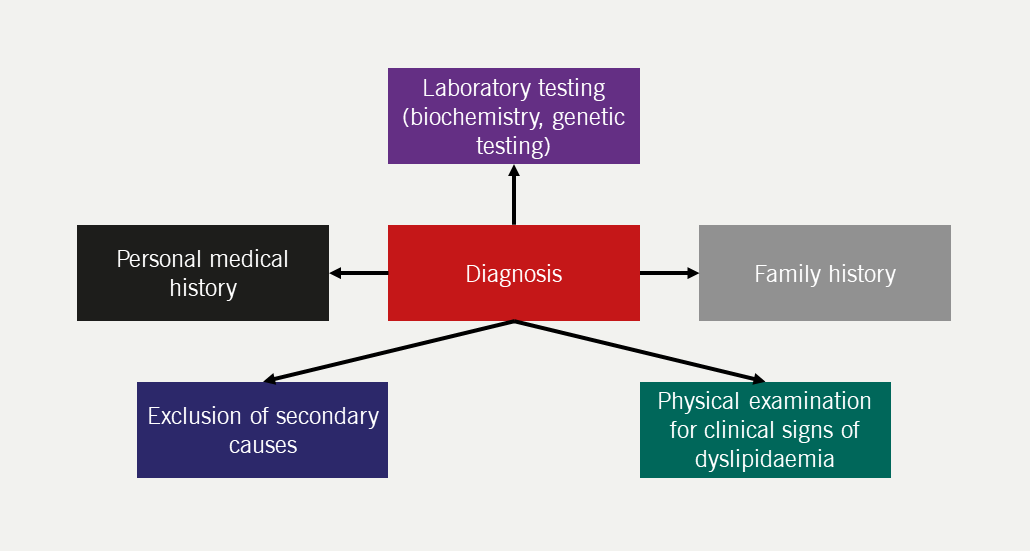
Accurate diagnosis is dependent upon careful clinical assessment (figure 1), and early treatment can prevent major cardiovascular complications.
Secondary dyslipidaemias
Table 2. Key tests to exclude secondary causes of dyslipidaemia
| Disorder | Biochemical test |
| Diabetes mellitus | HbA1c |
| Chronic kidney disease | Renal profile (sodium, potassium, urea, creatinine, eGFR) |
| Nephrotic syndrome | Dipstick urinary protein |
| Liver failure | Liver profile (ALT, AST, GGT, albumin) |
| Hypothyroidism | Thyroid profile |
| Key: ALT = alanine transaminase; AST = aspartate transaminase ; eGFR = estimated glomerular filtration rate; GGT = gamma-glutamyl transpeptidase; HbA1c = glycated haemoglobin A1c | |
It is important to recognise secondary causes of dyslipidaemia because the treatment of the underlying condition should be the primary focus of management (table 2). Table 3 shows the pattern of lipoprotein disturbance according to the underlying secondary cause. In some cases, it may be pathognomonic, such as the presence of lipoprotein X (LpX) which is a lipoprotein formed in cholestatic conditions and often erroneously reported as low-density lipoprotein cholesterol (LDL-C). A low apolipoprotein B (apoB) can support the diagnosis of LpX in patients with cholestatic disease.
Table 3. Possible changes in lipoprotein levels seen in secondary causes of dyslipidaemia4,5
| Disorder | Lipoprotein disturbance | |||
| ↑ LDL-C | ↓ HDL-C | ↑ Triglycerides | ↑ Lp(a) | |
| Acromegaly | ✓ | ✓ | ✓ | ✓ |
| Alcohol excess | – | ↑ | ✓ | – |
| Anorexia nervosa | ✓ | – | – | – |
| Cholestasis | ✓ | – | – | – |
| Chronic kidney disease | – | – | ✓ | ✓ |
| Cushing’s syndrome | ✓ | ✓ | ✓ | – |
| Hyperthyroidism | ↓ | ✓ | ✓ | – |
| Hypothyroidism | ✓ | ↑ | ✓ | ✓ |
| Menopause | ✓ | ✓ | – | ✓ |
| Nephrotic syndrome | ✓ | – | ✓ | ✓ |
| Obesity | ✓ | ✓ | ✓ | – |
| Polycystic ovary syndrome | ✓ | ✓ | ✓ | – |
| Type 1 or 2 diabetes mellitus | ✓ | ✓ | ✓ | – |
| Key: HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; Lp(a) = lipoprotein(a) | ||||
Medications can also cause dyslipidaemia with variable patterns of lipoprotein disturbance according to the class of medication (table 4).
Table 4. Medications that can cause dyslipidaemia and their pattern of disturbance
| Class of medication | Lipoprotein disturbance |
| Atypical antipsychotics Corticosteroids Ciclosporin |
↑ Cholesterol and triglyceride levels |
| Beta blockers HIV / antiretroviral drugs Oestrogens Retinoids |
↑ Triglyceride levels |
| Anabolic steroid | ↓ HDL-C levels |
| Key: HDL-C = high-density lipoprotein cholesterol; HIV = human immunodeficiency virus | |
Take a look at case scenario 1 in the box below, which illustrates the importance of considering secondary causes of dyslipidaemia.

| This is not the image of the case, and is used for illustrative purposes |
Case scenario 1
Consider secondary causes of dyslipidaemia
Carol, a 49-year-old teaching assistant, was referred to the lipid clinic following admission with acute chest pain (subsequently considered non-cardiac, cardiac troponin negative). Her total cholesterol at the time of admission was 10.0 mmol/L (optimal: <5 mmol/L). She had previously undergone thyroidectomy and radio-iodine ablation for papillary thyroid cancer and was therefore at risk of hypothyroidism. She had a history of hypertension (blood pressure [BP] 126/80 mmHg), was overweight (body mass index [BMI] 29.0 kg/m2), physically active, a moderate drinker (10 units per week), an ex-smoker, and dietary assessment indicated scope for improvement. Both parents had type 2 diabetes mellitus (DM) and hypercholesterolaemia but no clinical cardiovascular disease (CVD). She was discharged on atorvastatin 40 mg/day, aspirin 75 mg/day, levothyroxine 125 μg/day and lisinopril 5 mg/day. There was no evidence of stigmata associated with hyperlipidaemia.
At the clinic one month later, her total cholesterol was 5.2 mmol/L (LDL-C: 3.7 mmol/L [optimal: <2.6 mmol/L], triglycerides: 1.3 mmol/L [optimal: <1.69 mmol/L], high-density lipoprotein cholesterol [HDL-C]: 1.2 mmol/L [optimal: >1.0 mmol/L for men, >1.2 mmol/L for women]) and thyroid profile was within the target range for suppressive treatment, given her history of papillary thyroid cancer (thyroid stimulating hormone [TSH] <0.05 mU/L [normal range: 0.4–4.0 mU/L], free thyroxine [FT4]: 25.2 pmol/L [normal range: 9.0–25.0 pmol/L]), on levothyroxine therapy. As post-ablation hypothyroidism was considered a possible cause of her dyslipidaemia when she was admitted to hospital with chest pain, atorvastatin was withdrawn to reassess her lipid profile.
Repeat lipid testing one month later showed a total cholesterol of 5.3 mmol/L (LDL-C: 3.5 mmol/L, triglycerides: 1.5 mmol/L and HDL-C: 1.0 mmol/L). Her QRISK®3 10-year cardiovascular risk was estimated at 8%. Carol was advised to continue with therapeutic lifestyle interventions and return for repeat lipid testing in six months.
Learning point
This case scenario highlights the importance of considering secondary causes of dyslipidaemia before initiating lipid-modifying treatment. Carol’s hypothyroidism due to radio-iodine ablation was the probable cause of dyslipidaemia. Following commencement of levothyroxine, her TSH and FT4 levels had normalised, and her cholesterol had returned to more normal levels, which were maintained after stopping statin therapy.
Familial hypercholesterolaemia
Prevalence
In the absence of secondary causes, a strong family history of premature CHD is suggestive of an atherogenic familial lipid disorder. Of all the inherited causes of dyslipidaemia, familial hypercholesterolaemia (FH) is the most widely recognised, with an estimated prevalence of the heterozygous form in Caucasians of one in 250 (0.4%).6 This is twice as high as previously thought and suggests that in the UK, between 130,000 to 260,000 people have FH,7 but only around 20,000 or so cases have been identified. FH is present from birth and almost all affected people are heterozygotes (HeFH). Homozygous FH (HoFH) is extremely rare with a prevalence of one in one million8 (see table 5).
Table 5. Prevalence of common genetic dyslipidaemias in people of European origin
| Disorder | Abnormal lipids | Prevalence |
| Familial combined hyperlipidaemia (FCH) | ↑ LDL-C, ↑ triglycerides, ↑ total cholesterol | 1:100 |
| Heterozygous familial hypercholesterolaemia (HeFH) | ↑ LDL-C (typical range 5–10 mmol/L in HeFH and 10–20 mmol/L in HoFH) ↑ apoB |
1:250 |
| Homozygous familial hypercholesterolaemia (HoFH) | 1:1,000,000 | |
| Polygenic hypercholesterolaemia | 1:50 | |
| Familial hypertriglyceridaemia | ↑ VLDL and ↑ triglycerides | 1:100 |
| Key: apoB = apolipoprotein B; FCH = familial combined hyperlipidaemia; HeFH = heterozygous familial hypercholesterolaemia; HoFH = homozygous familial hypercholesterolaemia; LDL-C = low-density lipoprotein cholesterol; VLDL = very-low-density lipoprotein | ||
FH markedly elevates serum LDL-C levels and, if untreated leads to premature atherosclerotic cardiovascular disease (ASCVD). LDL-C levels are elevated from birth, reaching 5–10 mmol/L in adulthood.9 The presence of two pathogenic alleles in HoFH can increase serum LDL-C to ten times the mean concentration for age- and gender-matched individuals.10 The much rarer HoFH and compound heterozygous FH should be suspected in individuals with a pre-treatment LDL-C greater than 13 mmol/L in adults and 11 mmol/L in children.1,2,10
HeFH increases the risk of premature CHD4 by over 50% in men and at least 30% in women.11 If HeFH is not treated, 50% of men and 15% of women will have developed symptomatic CHD by the age of 50,2,12 but with effective lipid-lowering therapy, individuals with HeFH can achieve a normal life expectancy.4 In HoFH, however, severe aorto-coronary disease may be found in childhood and as response to lipid-lowering therapy is poor, lipoprotein apheresis is required.13
Causes of FH
FH is primarily an inherited autosomal co-dominant monogenic disorder affecting the LDL-receptor pathway.16 Variants in genes encoding the LDL-receptor (LDLR), apolipoprotein (apo) B100 (the LDL-receptor ligand) and proprotein convertase subtilisin/kexin type 9 (PCSK9) reduce hepatic uptake and degradation of LDL-C from the systemic circulation.16 It is estimated that >90% of FH causative mutations are attributable to LDLR, 5% due to APOB and 1% to PCKS9.8,17 Further advances have revealed autosomal recessive mutations leading to loss of function in LDL receptor adaptor protein 1 (LDLRAP1) and very rare heterozygous mutations in APOE.18 Although other undiscovered genes may account for some cases of monogenic FH, the absence of a mutation suggests polygenic hypercholesterolaemia, which does not usually show a clear dominant pattern of inheritance.
Clinical diagnosis
Two sets of diagnostic criteria have been designed to determine the likelihood of FH; the Simon Broome Criteria17 (table 6) and the Dutch Lipid Network Criteria (DLNC)18 (table 7).
Table 6. Simon Broome diagnostic criteria for FH in adults17
| Simon Broome diagnostic criteria for FH |
|---|
| Adult: total cholesterol >7.5 mmol/L or LDL-C >4.9mmol/L Children: total cholesterol >6.7 mmol/L or LDL-C >4.0 mmol/L |
| Definite FH |
Plus at least one of the following two:
OR |
| Possible FH |
Plus at least one of the following two:
OR |
| Key: APOB = apolipoprotein B; FH = familial hypercholesterolaemia; LDL-C = low-density lipoprotein cholesterol; LDLR = low-density lipoprotein receptor; PCSK9 = proprotein convertase subtilisin/kexin type 9 |
Table 7. Dutch Lipid Clinic Network diagnostic criteria for FH18
| Dutch Lipid Clinic Network diagnostic criteria for FH | Score |
| I. Family history | |
| First-degree relative with known premature coronary and vascular disease (<55 years male; <60 years female) OR First-degree relative with known LDL-C above the 95th percentile |
1 |
| First-degree relative with tendon xanthomas and/or corneal arcus OR Children <18 years with known LDL-C above the 95th percentile |
2 |
| II. Clinical history | |
| Premature coronary artery disease (<55 years male; <60 years female) | 2 |
| Premature cerebral or peripheral vascular disease (<55 years male; <60 years female) | 1 |
| III. Physical examination | |
| Tendon xanthomas | 6 |
| Corneal arcus (<45 years) | 4 |
| IV. LDL-C level (mmol/L) | |
| >8.5 | 8 |
| 6.5–8.4 | 5 |
| 5.0–6.4 | 3 |
| 4.0–4.9 | 1 |
| V. DNA analysis | |
| Functional mutation in the LDLR, APOB or PCSK9 genes | 8 |
| FH diagnosis | Total |
|---|---|
| Definite | >8 |
| Probable | 6–8 |
| Possible | 3–5 |
| Unlikely | 0–2 |
| Key: APOB = apolipoprotein B; FH = familial hypercholesterolaemia; LDL-C = low-density lipoprotein cholesterol; LDLR = low-density lipoprotein receptor; PCSK9 = proprotein convertase subtilisin/kexin type 9 | |
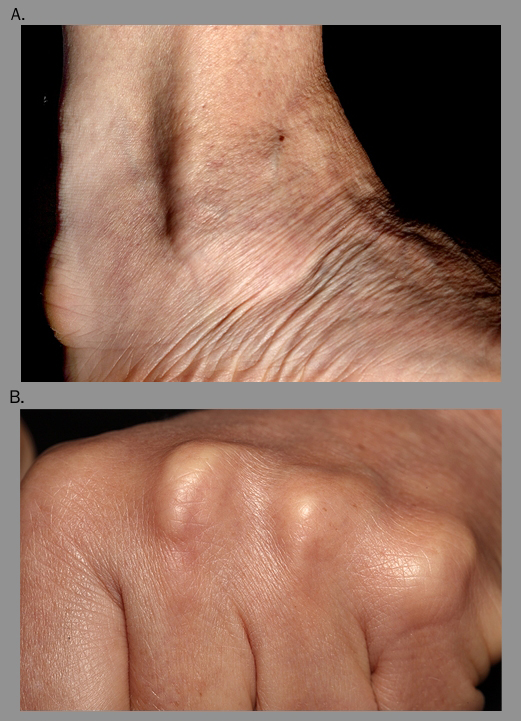
| Pictures reproduced with kind permission from Dermot Neely |
The finding of tendon xanthomas is virtually pathognomonic and confirms the clinical diagnosis of definite FH according to the Simon Broome criteria (table 6). Tendon xanthomas are subcutaneous nodules that slowly enlarge and arise from cholesterol deposits attached to tendons, most commonly the Achilles tendon or over the knuckles (figure 2). Tendon xanthomas suggest a significantly increased risk of CVD across all age groups.19 Tendon xanthomas are found in fewer than 30% of cases, most of whom have monogenic FH with a disease-defining mutation in LDLR, APOB or PCSK9 genes.2
Clinical characteristics (physical signs, cardiovascular risk factors and/or disease) observed in adults with FH are uncommon in children and adolescents with FH. Detection in this age group relies on measurement of LDL-C and, where available, genetic confirmation.20,21
Underdiagnosis
In 2021, the European Atherosclerosis Society (EAS) Familial Hypercholesterolaemia Studies Collaboration reported that in adults with FH who were diagnosed between the age of 40–49 years, more than one in six adults had established ASCVD. Only 2.1% of adults with FH were diagnosed in childhood or adolescence22 and it is estimated that undiagnosed FH might be responsible for one in 10 myocardial infarctions under 50 years of age.23
Healthcare systems worldwide identify <10% of individuals of any age with FH and in the UK only 8–15% of FH cases are known.11,22 As one child is born with FH every minute,24 approaches to early detection should be revised to reduce the deficit between prevalence and detection.
Primary care can aid in the diagnosis of FH as the average group practice of 10,000 patients will have around 16 cases of FH. Although it sounds proportionately low, early initiation of lipid-lowering therapy may substantially mitigate the risk of premature ASCVD and cardiac death.25
The National Institute for Health and Care Excellence (NICE) recommends the systematic identification of at-risk patients within primary care for onward referral, genetic testing and cascade testing (identifying people at risk for a genetic condition by tracing it through their family).2 Additionally, NICE recommends DNA analysis for the confirmation of FH in the index case and family cascade testing where the mutation has been identified.2 Genetic testing in FH has several advantages, such as improved precision of diagnosis, counselling, risk stratification, adherence to treatment, access to specialist therapies and the cost-effectiveness of cascade testing. If no mutation is identified in the index case or genetic testing is not available, affected relatives can be identified on the basis of age- and sex-specific LDL-C thresholds (as recommended by NICE); however, in up to one-third of cases, it may not be possible to make a firm diagnosis, making this approach much less efficient.2


Great strides are being made by HEART UK – The Cholesterol Charity and the British Heart Foundation (BHF) to increase awareness of FH. Both charities produce useful and practical booklets on the condition:
HEART UK – ‘Familial hypercholesterolaemia: An educational booklet for people with familial hypercholesterolaemia (FH)’
British Heart Foundation – ‘Life with familial hypercholesterolaemia’
A nationwide, proactive, systematic approach to cascade is recommended in guidelines,2,5 but commissioning support for implementation is lacking in most parts of the UK.
National FH services have been established in Northern Ireland, Scotland and Wales. The National health Service (NHS) Long Term Plan in 2019 aims to improve the identification rate of FH cases to at least 25% in the course of five years through the NHS Genomics programme.26 As of April 2020, genetic testing for FH is centrally funded in England through the Genomics England programme as a means to improve detection rates. The use of cascade screening is cost effective and enables affected individuals to be identified from childhood, which not only lowers their lifetime risk, but also contributes to reducing CVD burden at the population level.11
Combined hyperlipidaemia
A mixed lipid profile showing raised total cholesterol, triglycerides or both can be suggestive of a number of dyslipidaemias, including:
- familial combined hyperlipidaemia (FCH)
- remnant hyperlipidaemia (type III or familial dysbetalipoproteinaemia)
- dyslipidaemia associated with the metabolic syndrome
- milder presentations of familial hypertriglyceridaemia.
All appear to share a common genetic basis, with an increased burden of common triglyceride-raising gene variants conferring susceptibility to adverse lifestyle and secondary causes of dyslipidaemia, such as obesity.
Table 8. Lipid profile associated with FCH3
| ↑ Total cholesterol (6.5–10.0 mmol/L) |
| ↑ LDL-C |
| ↑ Triglycerides (2.3–6.0 mmol/L or higher) |
| ApoB/total cholesterol >0.15 |
| VLDL-C/total triglycerides <0.69* |
| Small, dense, LDL |
| Diagnosis is commonly based on the combination of apoB >1.20 g/L + triglycerides >1.5 mmol/L with a family history of premature cardiovascular disease2 |
| Key: apoB = apolipoprotein B; FCH = familial combined hyperlipidaemia; LDL = low-density lipoprotein; LDL-C = low-density lipoprotein cholesterol; VLDL-C = very-low-density lipoprotein cholesterol * Determined by ultracentrifugation |
FCH is a common metabolic disorder and affects about one in 100 people.1 The underlying mechanism involves overproduction of very-low-density lipoprotein (VLDL) and apoB. The genetic basis is complex and influenced by environmental factors. As there is considerable variability in presentation, diagnosis can often be missed in practice. FCH should be suspected if total cholesterol levels are between 6.5 and 9.0 mmol/L and/or triglycerides between 2.3 and 5.0 mmol/L (see table 8).
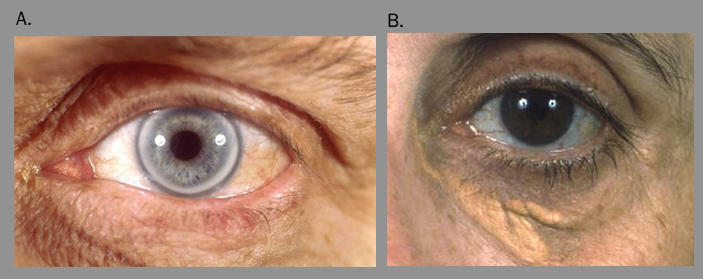
Elevated levels of cholesterol and triglyceride, either alone or in combination, in patients and other family members confer a ‘variable phenotype’. FCH has many overlapping features with the metabolic syndrome which can make differential diagnosis challenging in clinical practice (figure 3).27 ApoB is invariably elevated; therefore, a useful diagnostic tool is to strongly consider FCH when apoB levels >1.20 g/L together with elevated triglycerides and family history of CVD.13 The finding of an apoB concentration that is unexpectedly low (apoB/total cholesterol ratio <0.15 g/mmol) raises the suspicion of remnant hyperlipidaemia (type III or familial dysbetalipoproteinaemia).28 The presence of xanthelasma (figure 4) is not of specific diagnostic significance but is more frequently seen in FCH and represents an area of lipid-laden macrophages, which is independently predictive of an increased risk of CHD, atherosclerosis and mortality.29

| Key: ApoB = apolipoprotein B; CVD = cardiovascular disease; FCH = familial combined hyperlipidaemia; HDL = high-density lipoprotein; LDL = low-density lipoprotein |
| Pictures reproduced with kind permission from Dermot Neely |
Case scenario 2 in the box below describes how to differentiate familial causes of combined hyperlipidaemia.

| This is not the image of the case, and is used for illustrative purposes |
Case scenario 2
Differentiating familial causes of combined hyperlipidaemia
Derek, a 40-year-old former RAF engineer, was referred to the lipid clinic after recent admission to the rapid-access chest pain clinic with burning central chest pain (exercise electrocardiogram [ECG] was negative). Atorvastatin was initiated by the clinician but Derek later stopped this due to severe headache and fatigue.
Derek was a non-smoker, physically active, a light drinker (4–6 units per week), was overweight (BMI 29.5 kg/m2) and had well-controlled BP (136/84 mmHg), and normal renal, hepatic and thyroid function. Fasting glucose was 4.7 mmol/L (normal: 3.9–5.5 mmol/L). There was no family history of CVD. However, there was evidence of xanthelasma together with an elevated fasting total cholesterol level of 8.1 mmol/L (optimal: <5 mmol/L), although triglycerides were near normal at 1.6 mmol/L (optimal: <1.7 mmol/L). On a repeat fasting test one month later, both were higher with total cholesterol of 8.4 mmol/L and triglycerides of 3.4 mmol/L. HDL-C was within normal limits at 1.4 mmol/L (optimal: >1.0 mmol/L for males, >1.2 mmol/L for females).
Learning point
This case scenario highlights the variable phenotype of the lipid abnormality in combined dyslipidaemia and the limitations of family history. The first profile is suggestive of possible FH, the second suggests that a diagnosis of combined dyslipidaemia typically associated with metabolic syndrome may be more likely. At least two lipid profiles are required to make a diagnosis.
Severe hypertriglyceridaemia
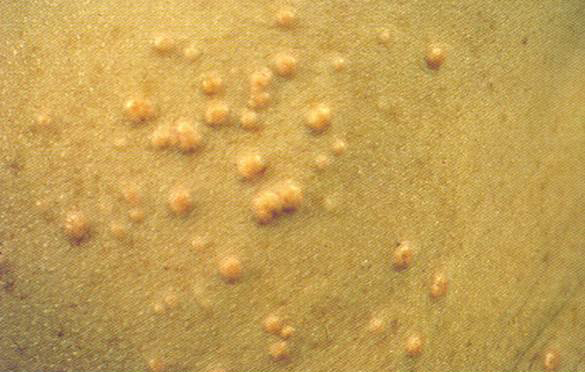
Patients with severely elevated triglyceride levels (>10 mmol/L) have significant accumulation of chylomicrons in the fasting state and are at a greatly increased risk of pancreatitis in a dose-dependent manner (figure 5).
| Picture reproduced with kind permission from Dermot Neely |
Like those with mild to moderate hypertriglyceridaemia, patients with severe hypertriglyceridaemia also have underlying polygenic defects impacting triglyceride clearance with additional secondary precipitating factors (tables 3 and 4). Elevated triglyceride levels are commonly associated with obesity, metabolic syndrome and DM as well as certain lifestyle factors, such as excessive alcohol intake, and is causally linked to atherosclerosis. Treatment includes lifestyle modification such as abstinence from alcohol, as well as exercise, weight loss, blood glucose control, and administration of fibrates and omega-3 fatty acids.30
However, those presenting with severe disease at a young age or recurrent pancreatitis in childhood are more likely to have an underlying monogenic cause. A rare familial cause of severe hypertriglyceridaemia is familial chylomicronaemia syndrome, which has an autosomal recessive inheritance and can present in childhood.31 Once secondary causes have been addressed, dietary fat restriction is the cornerstone of management. Treatment with statins may be ineffective and most patients with predominant hypertriglyceridaemia respond better to fibrate medications.
Lipid profiles
Clearly the lipid profile is key to the diagnosis of dyslipidaemia, and its key components are summarised in figure 6. The baseline lipid evaluation should comprise total cholesterol, triglycerides and HDL-C.2 Because total cholesterol involves the measurement of both atherogenic (LDL-, intermediate-density lipoprotein [IDL]- and VLDL-C) and anti-atherogenic (HDL-C) lipid fractions, it is inadequate for monitoring treatment. Instead, LDL- and non-HDL-C are preferred; non-HDLC represents the total sum of cholesterol in apoB-containing lipoproteins (chylomicron remnants, VLDL, IDL, LDL, lipoprotein[a] [Lp(a)]), which are atherogenic.
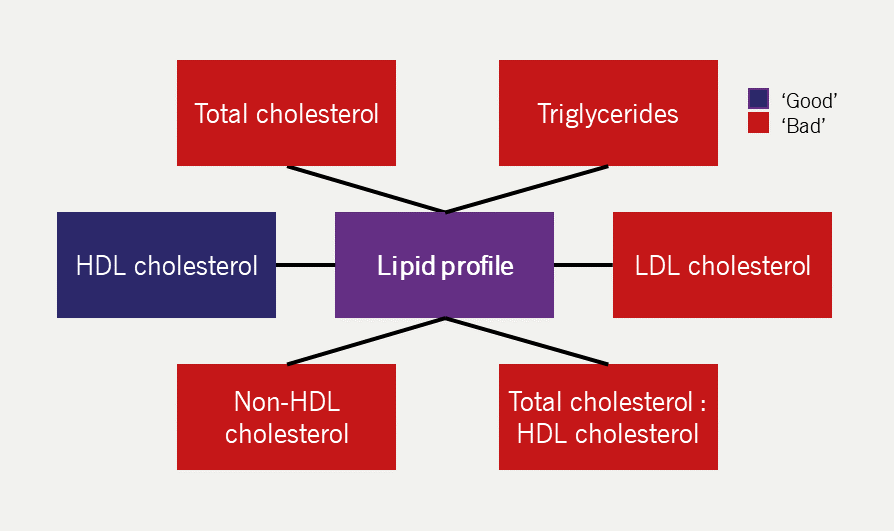
| Key: HDL = high-density lipoprotein; LDL = low-density lipoprotein |
Of the two, there is a strong case for preferential use of non-HDL-C, given that:
- it is a simple calculation (non-HDL-C = total cholesterol – HDL-C)
- it can be readily measured in non-fasting samples32
- it captures all atherogenic apoB-containing lipoproteins (VLDL, IDL, LDL, chylomicron remnants and Lp[a]).
In contrast, LDL-C is widely calculated by the Friedewald equation, which requires fasting samples. It is calculated as follows:
LDL-C = TC – (HDL-C + TG/2.2) [In mmol/L]
| Key: HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; TC = total cholesterol; TG = triglyceride |
The Friedewald equation was developed in 1972 for research purposes from a population of 448 individuals and has been widely adopted in clinical practice for several decades.33 The Friedewald equation assumes a constant cholesterol to triglyceride ratio in VLDL of 0.45; however, this assumption does not hold true in non-fasting conditions or when the fasting triglyceride concentration exceeds 4.5 mmol/L.2 Furthermore, the ratio is altered by statin treatment. Therefore, the main role for calculated LDL-C is in the assessment of suspected FH before initiating treatment. In clinical practice, LDL-C levels have a role in monitoring response to lipid-lowering treatment as well as setting recommended target levels in FH patients and those with or without pre-existing CVD as per the 2019 European Society of Cardiology (ESC) /EAS guidelines (see module 4).1 LDL-C concentration is also used in the NICE technology appraisal (TA394) guidance as a threshold for offering treatment with monoclonal antibodies and small interfering RNA to PCSK9 inhibitors.34
LDL-C concentration can also be estimated using the Sampson equation:35
LDL-C = TC/0.948 − HDL-C/0.971 − (TG/8.56 + TG × non-HDL-C/2140 − TG2/16100) − 9.44
| Key: HDL-C = high-density lipoprotein cholesterol; LDL-C = low-density lipoprotein cholesterol; nHDL-C = non-high-density lipoprotein cholesterol; TC = total cholesterol; TG = triglyceride |
The Sampson equation was developed in 2020 using 18,715 lipid samples from 8,656 patients.35 The Sampson equation has several advantages compared to the Friedewald equation:
- Better accuracy at low LDL-C concentrations
- LDL-C can be determined in samples containing triglycerides up to a concentration of 9 mmol/L
- Validated in non-fasting samples.
Therefore, the Sampson equation displays improved performance compared to the Friedewald equation.35
Lp(a)
Table 9. Cardiovascular risk as determined by Lp(a) serum concentration, as per the HEART UK consensus statement40
| Lp(a) level (nmol/L) | Cardiovascular risk |
| 32–90 | Minor |
| 90–200 | Moderate |
| 200–400 | High |
| >400 | Very high |
| Key: Lp(a) = lipoprotein(a) | |
Table 10. Patient groups in whom the HEART UK consensus statement recommends Lp(a) measurement40
| Lp(a) measurement is recommended in: |
|---|
| a. A personal or family history of premature ASCVD (<60 years) |
| b. First-degree relatives with a raised Lp(a) level (>200 nmol/L) |
| c. Genetic dyslipidaemias such as FH |
| d. Calcific aortic valve stenosis |
| e. Borderline increased (<15%) 10-year risk of a cardiovascular event |
| Key: ASCVD = atherosclerotic cardiovascular disease; FH = familial hypercholesterolaemia; Lp(a) = lipoprotein(a) |
| With kind permission from Royal Brompton & Harefield NHS Foundation Trust |
Lp(a) is an atherogenic and prothrombotic LDL-like particle, that promotes atherosclerosis.36 Lp(a) levels are primarily genetically determined with an autosomal co-dominant inheritance.37 The EAS recommends Lp(a) measurement in all adults at least once during the lifetime unless a secondary cause is suspected (table 4), or a specific treatment is instituted to lower levels.38 Serum Lp(a) has become established as an independent and causal risk factor for ASCVD and a new risk factor for calcific aortic valve stenosis38 as Lp(a) is associated with an increased risk of CVD, particularly if levels exceed 125 nmol/L (50 mg/dL or 500 mg/L) where the risk of myocardial infarction is increased two- to threefold.11 The association between elevated Lp(a) and increased CVD risk appears continuous and independent of LDL-C levels across different ethnic groups.39 As such, the HEART UK consensus statement on Lp(a) recommends its measurement in five main categories which identifies at-risk patients for cardiovascular events (tables 9 and 10).40 There is no specific treatment available to lower Lp(a) currently, apart from lipoprotein apheresis (figure 7), but clinical trials for novel agents are in development. In clinical practice, aspirin can be used to mitigate the prothrombotic risk associated with an elevated Lp(a) and hyperlipidaemic control is recommended with a non-HDL-C target of <2.5 mmol/L.40
Although there has been much debate concerning the role of other novel risk factors such as the systemic inflammatory biomarker C-reactive protein (CRP), measured by a high-sensitivity CRP assay (hsCRP), the currently available evidence suggests that these add little and are not recommended for use in routine assessment of CVD risk in UK guidelines (NICE NG2382 and Joint British Societies for the Prevention of Cardiovascular Disease [JBS3]36). A recent study has shown that regardless of baseline hsCRP value, higher levels of Lp(a) were associated with major adverse cardiac events, myocardial infarction and peripheral arterial disease in both primary (UK Biobank) and secondary prevention populations (FOURIER [Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk] and SAVOR-TIMI 53 [Saxagliptin Assessment of Vascular Outcomes Recorded in Patients with Diabetes Mellitus-Thrombolysis in Myocardial Infarction 53] clinical trials).41
Summary
Dyslipidaemias can be considered a primary lipid disorder or secondary to an underlying disease or medication. Known secondary causes of dyslipidaemia can be identified on clinical assessment and it is important to treat the underlying cause. Familial forms of dyslipidaemia are common and are underdiagnosed and undertreated. In particular, FH is associated with elevated total and LDL-C levels and greatly increases the risk of premature coronary artery disease. More rare and severe causes of dyslipidaemia, such as HoFH, can cause advanced aorto-coronary atherosclerosis earlier in life and often require lipoprotein apheresis. Combined hyperlipidaemia arises where elevations of triglyceride are seen in tandem with elevations in total and LDL-C. Hypertriglyceridaemias have mixed monogenic or polygenic aetiology and place patients at risk of developing pancreatitis when severe.
Key messages
- The diagnosis of dyslipidaemia should involve a thorough clinical assessment and secondary causes of dyslipidaemia should be excluded
- LDL-C is important in the diagnosis of FH; apoB is diagnostic in mixed dyslipidaemia
- Measurement of Lp(a) should be considered in patients with premature CHD.
Recommended reading:
Watts GF, Gidding S, Wierzbicki AS et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol 2014;171:300–25. http://dx.doi.org/10.1016/j.ijcard.2013.11.025 [Epub online ahead of print]
close window and return to take test
References
- Mach F, Baigent C, Catapano AL et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J 2020;41:111–88. https://doi.org/10.1093/eurheartj/ehz455
- National Institue for Health and Care Excellence. Cardiovascular disease: risk assessment and reduction, including lipid modification [NG238]. London: NICE, published date: 14 December 2023. Available at: www.nice.org.uk/guidance/ng238 (accessed March 2024)
- Genest JJ Jr, Martin-Munley SS, McNamara JR et al. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation 1992;85:2025–33. https://doi.org/10.1161/01.cir.85.6.2025
- Cegla J, Scott J. Lipid disorders. In: Oxford Textbook of Medicine, 6th Edition. Oxford: Oxford University Press, 2020. Available at: https://oxfordmedicine.com/mobile/view/10.1093/med/9780198746690.001.0001/med-9780198746690-chapter-232
- Newman CB, Blaha MJ, Boord JB et al. Lipid management in patients with endocrine disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2020;105:dgaa674. https://doi.org/10.1210/clinem/dgaa674. Erratum in: J Clin Endocrinol Metab 2021;106:e2465.
- Akioyamen LE, Genest J, Shan SD et al. Estimating the prevalence of heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. BMJ Open 2017;7:e016461. https://doi.org/10.1136/bmjopen-2017-016461
- Versmissen J, Oosterveer DM, Yazdanpanah M et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008;337:a2423. https://doi.org/10.1136/bmj.a2423
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004;160:407–20. http://doi.org/10.1093/aje/kwh236
- Watts GF, Gidding SS, Hegele RA et al. International Atherosclerosis Society guidance for implementing best practice in the care of familial hypercholesterolaemia. Nat Rev Cardiol 2023;20:845–69. https://doi.org/10.1038/s41569-023-00892-0
- France M, Rees A, Datta D et al. HEART UK statement on the management of homozygous familial hypercholesterolaemia in the United Kingdom. Atherosclerosis 2016;255:128–39. https://doi.org/10.1016/j.atherosclerosis.2016.10.017 [Epub online ahead of print]
- Public Health England (PHE). Familial hypercholesterolaemia: implementing a systems approach to detection and management. London: PHE, 2018. Available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/731873/familial_hypercholesterolaemia_implementation_guide.pdf (accessed March 2024)
- Ray KK, Pillas D, Hadjiphilippou S et al. Premature morbidity and mortality associated with potentially undiagnosed familial hypercholesterolemia in the general population. Am J Prevent Cardiol 2023;15:100580. https://doi.org/10.1016/j.ajpc.2023.100580 [Epub online ahead of print]
- Nordestgaard BG, Chapman MJ, Ray K et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J 2010;31:2844–53. https://doi.org/10.1093/eurheartj/ehq386
- Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med 2007;4:214–25. https://doi.org/10.1038/ncpcardio0836
- Nordestgaard BG, Chapman MJ, Humphries SE et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013;34:3478–90. https://doi.org/10.1093/eurheartj/eht273 [Epub online ahead of print]
- Iacocca MA, Hegele RA. Recent advances in genetic testing for familial hypercholesterolemia. Expert Rev Mol Diagn 2017;17:641–51. https://doi.org/10.1080/14737159.2017.1332997 [Epub online ahead of print]
- Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 1991;303:893–6. https://doi.org/10.1136/bmj.303.6807.893
- WHO Human Genetics Programme. Familial hypercholesterolaemia (FH): report of a second WHO consultation, Geneva, 4 September 1998. Geneva: World Health Organisation, 1999. Available at: https://apps.who.int/iris/handle/10665/66346 (accessed March 2024)
- Civeira F, Castillo S, Alonso R et al. Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the low-density lipoprotein receptor gene mutation. Arterioscler Thromb Vasc Biol 2005;25:1960–5. https://doi.org/10.1161/01.ATV.0000177811.14176.2b [Epub online ahead of print]
- European Atherosclerosis Society Familial Hypercholesterolaemia Studies Collaboration. Familial hypercholesterolaemia in children and adolescents from 48 countries: a cross-sectional study. Lancet 2024;403:55–66. https://doi.org/10.1016/S0140-6736(23)01842-1 [Epub online ahead of print]
- Gidding SS. Childhood screening for familial hypercholesterolemia: JACC Review Topic of the Week. J Am Coll Cardiol 2023;82:1558–63. https://doi.org/10.1016/j.jacc.2023.07.028
- Vallejo-Vaz AJ, Stevens CA, Lyons AR et al. Global perspective of familial hypercholesterolaemia: a cross-sectional study from the EAS Familial Hypercholesterolaemia Studies Collaboration (FHSC). Lancet 2021;398:1713–25. https://doi.org/10.1016/S0140-6736(21)01122-3 [Epub online ahead of print]
- National Organization for Rare Disorders. Familial hypercholesterolemia. 2023. https://rarediseases.org/rare-diseases/familial-hypercholesterolemia/ (accessed December 2023)
- Wiegman A, Gidding SS, Watts GF et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J 2015;36:2425–37. https://doi.org/10.1093/eurheartj/ehv157 [Epub online ahead of print]
- Durand A, Morgan CLI, Tinsley S, Hughes E, McCormack T, Bitchell CL, Lahoz R. Familial hypercholesterolaemia in UK primary care: a Clinical Practice Research Datalink study of an under-recognised condition. Br J Gen Pract 2024;74:e174–e82. https://doi.org/10.3399/BJGP.2023.0010 [Epub online ahead of print]
- National Health Service. The NHS Long Term Plan 2019. Available at: www.longtermplan.nhs.uk (accessed March 2024)
- Gaddi A, Cicero AF, Odoo FO et al. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag 2007;3:877–86.
- Blom DJ, O’Neill FH, Marais AD. Screening for dysbetalipoproteinemia by plasma cholesterol and apolipoprotein B concentrations. Clin Chem 2005;51:904–7. https://doi.org/10.1373/clinchem.2004.047001
- Christoffersen M, Frikke-Schmidt R, Schnohr P, Jensen GB, Nordestgaard BG, Tybjaerg-Hansen A. Xanthelasmata, arcus corneae, and ischaemic vascular disease and death in general population: prospective cohort study. BMJ 2011;343:d5497. https://doi.org/10.1136/bmj.d5497
- Parhofer KG, Laufs U. The diagnosis and treatment of hypertriglyceridemia. Dtsch Arztebl Int 2019;116:825–32. https://doi.org/10.3238/arztebl.2019.0825
- Durrington P. Dyslipidaemia. Lancet 2003;362:717–31. https://doi.org/10.1016/S0140-6736(03)14234-1
- Di Angelantonio E, Sarwar N, et al. The Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009;302:1993–2000.
- Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499–502.
- National Institue for Health and Care Excellence. Evolocumab for treating primary hypercholesterolaemia and mixed dyslipidaemia. Technology appraisal [TA394]. London: NICE, published: 22 June 2016. www.nice.org.uk/guidance/ta394
- Sampson M, Ling C, Sun Q et al. A new equation for calculation of low-density lipoprotein cholesterol in patients with normolipidemia and/or hypertriglyceridemia. JAMA Cardiol 2020;5:540–8. https://doi.org/10.1001/jamacardio.2020.0013. Erratum in: JAMA Cardiol 2020;5:613.
- Gudbjartsson DF, Thorgeirsson G, Sulem P et al. Lipoprotein(a) concentration and risks of cardiovascular disease and diabetes. J Am Coll Cardiol 2019;74:2982–94. https://doi.org/10.1016/j.jacc.2019.10.019
- Kraft HG, Lingenhel A, Pang RW et al. Frequency distributions of apolipoprotein(a) kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: the distribution of null alleles is non-random. Eur J Hum Genet 1996;4:74–87. https://doi.org/10.1159/000472175
- Kronenberg F, Mora S, Stroes ESG et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J 2022;43:3925–46. https://doi.org/10.1093/eurheartj/ehac361 [Epub online ahead of print]
- Kronenberg F, Mora S, Stroes ESG, et al. Frequent questions and responses on the 2022 lipoprotein(a) consensus statement of the European Atherosclerosis Society. Atherosclerosis 2023;374:107–20. https://doi.org/10.1016/j.atherosclerosis.2023.04.012
- Cegla J, Neely RDG, France M, et al. HEART UK consensus statement on lipoprotein(a): A call to action. Atherosclerosis 2019;291:62–70. https://doi.org/10.1016/j.atherosclerosis.2019.10.011 [Epub online ahead of print]
- Small AM, Pournamdari A, Melloni GEM et al. Lipoprotein(a), C-reactive protein, and cardiovascular risk in primary and secondary prevention populations. JAMA Cardiol 2024;9:385–91. https://doi.org/10.1001/jamacardio.2023.5605 [Epub online ahead of print]

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.