A. Menarini Farmaceutica Internazionale SRL has provided an educational grant for the production of this e-learning programme and has had no editorial control or input. The views and content expressed within this programme are solely those of the authors.
PP-CA-UK-0151 Date of preparation: March 2020
Pathophysiology of angina
Angina pectoris is caused by myocardial ischaemia due to the reversible mismatch between myocardial oxygen supply and demand. This mismatch can occur during exercise, emotional stress or even spontaneously. Restriction of myocardial blood flow most commonly results from the narrowing of an epicardial (surface) coronary artery, usually from atherosclerosis, but other mechanisms may be involved, which may act independently or in combination. These mechanisms include:
- atherosclerosis (plaque-related epicardial coronary obstruction)
- focal or diffuse spasm of normal or atheromatous arteries
- microvascular dysfunction
- left ventricular dysfunction secondary to previous acute myocardial necrosis/hibernation (ischaemic cardiomyopathy)
- left ventricular hypertrophy (hypertension, aortic stenosis, hypertrophic cardiomyopathy).
A major determinant of myocardial ischaemia is increased heart rate,1 principally through increased myocardial oxygen demand and reduced diastolic perfusion. This provides the rationale for current therapeutic strategies in patients with angina pectoris (see Angina module 6: secondary prevention and treatment). In this module, we will focus on the most common mechanism for angina, namely epicardial coronary atherosclerosis.
Mechanisms of atherosclerosis
The pathophysiology of atherosclerosis, development of symptomatic angina and subsequent acute coronary syndrome (ACS) is well characterised.2,3 The microscopic development of atherosclerotic plaque involves:
- endothelial dysfunction
- sub-endothelial accumulation and oxidation of low-density lipoprotein (LDL)
- increased expression of adhesion molecules and chemokine secretion
- immune cell infiltration and development of foam cells
- development of fibroatheromatous plaque involving positive remodelling, neovascularisation, and development of necrotic core with overlying fibrous cap formation and calcification.
Endothelial dysfunction and nitric oxide
The endothelium is a single layer of cells lining blood vessels and performs multiple functions, including:
- the regulation of coronary blood flow
- a barrier to toxic substrates
- regulation of thrombosis and inflammation
- control of angiogenesis.
The principal mechanism by which the endothelium modulates coronary perfusion is by regulating coronary vascular tone through the production of nitric oxide (NO). This can be in response to a variety of stimuli, such as autonomic tone, shear stress, coronary driving pressure, inflammatory mediators, neuropeptides, temperature and neurohormonal responses.
NO is synthesised from the amino acid L-arginine by means of the enzyme endothelial NO synthase (e-NOS). Endothelial cells express e-NOS, producing a low level of NO, which is under the control of regulatory factors. NO has paracrine effects by readily permeating cell membranes where it activates the synthesis of cyclic guanylate cyclase (cGMP). Through this intracellular signalling pathway, it has a number of effects including vasodilation, angiogenesis, and reducing platelet adhesion and vascular smooth muscle proliferation.

Endothelial dysfunction is characterised by a reduction in the bioavailability of vasodilators, such as NO, as a result of reduced production and increased consumption of NO, and an increase in endothelium-derived contracting factors, such as endothelin and angiotensin II (AII) (figure 1). This is seen as the blood vessel being unable to dilate. In addition, endothelial dysfunction promotes platelet and leukocyte activation and the release of cytokines, which increase vessel permeability. This enables the sub-endothelial infiltration of oxidised lipoproteins and inflammatory cells.
Endothelial dysfunction has been referred to as “the risk of the risk factors”,4 since it has been shown to occur in the presence of conventional, modifiable risk factors (e.g. cigarette smoking, high blood pressure, hyperlipidaemia and diabetes). These risk factors are, in turn, associated with a state of increased oxidative stress and the overproduction of reactive oxygen species. This all contributes to the complex interplay of endothelial dysfunction since reactive oxygen species can themselves lead to abnormal NO metabolism (figure 2).
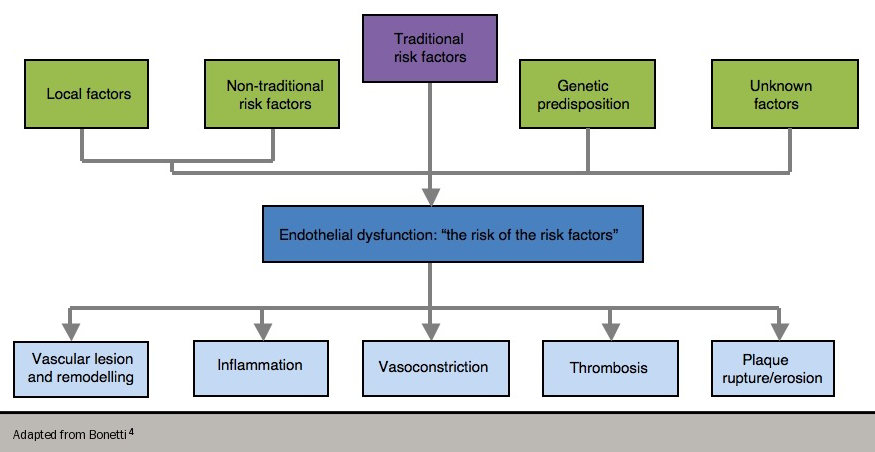
Consequently, endothelial dysfunction predisposes to atherogenesis and is a marker of atherosclerotic risk. A number of drugs that have been shown to reduce cardiovascular events, such as angiotensin-converting enzyme (ACE) inhibitors and statins, also improve endothelial dysfunction – this appears to play a fundamental role in the development of angina, and also in the more complex evolution of ACS.
The role of cigarette smoking in the development of atherosclerosis, angina and ACS has been extensively studied and reviewed.5,6 Smoking predisposes to atherogenesis through a number of processes including inflammation, platelet activation and fibrinolytic dysfunction, as well as modification of the lipid profile. In particular, smoking has been shown to cause altered vasomotor tone and impairs endothelium-dependent vasodilatation.7,8 Smoking causes a reduction in NO biosynthesis by reducing the expression and activity of e-NOS.9 There is limited evidence on other types of smoking but recent data suggest that electronic cigarettes and hookah (waterpipe) smoking are also associated with endothelial dysfunction.10,11
Furthermore, smoking is associated with increased expression of adhesion molecules and promotes increased platelet and macrophage adherence. Smoking has been shown to increase levels of multiple inflammatory markers including C-reactive protein (CRP), interleukin 6 (IL-6) and tumour necrosis factor (TNF-α), as well as increasing circulating leukocyte count in both male and female smokers by around 20–25%.12,13 Increased levels of oxidised low-density lipoprotein (LDL) cholesterol are also found in smokers – these particles have been shown to be actively taken up by macrophages to form foam cells.14 Cigarette smoking affects plaque stability and resultant ACS, as well as appearing to affect many aspects of atherogenesis and the spectrum of disease, from atherosclerosis development to angina and ACS. Smoking cessation is thus the most effective intervention for reversing cardiovascular risk and preventing poor outcomes.
Endothelial function can be assessed in a number of ways, though it should be noted that none are used routinely in clinical practice.15
Invasive assessment
Invasive assessment is carried out by the intra-arterial delivery of vasoactive agents (endothelium-dependent vasodilators, such as acetylcholine or substance P). Coronary endothelial function, vasodilation and changes in flow can be measured with high-resolution ultrasound, intracoronary Doppler flow wires or strain gauge plethysmography.
Acetylcholine activates endothelial cells and stimulates NO release, which physiologically causes vasodilatation and hyperaemia. When there is endothelial dysfunction, this process is impaired and in certain patients, there is a risk of paradoxical vasoconstriction and coronary ischaemia. Administering acetylcholine is invasive and costly.

Non-invasive assessment
Non-invasive assessment is carried out by:
- flow-mediated dilatory (FMD) capability of the brachial artery – a high-resolution vascular ultrasound investigation to find out the brachial artery response to reactive hyperaemia
- peripheral arterial tonometry (PAT) – an evaluation of pulse wave amplitude as a marker of endothelial dysfunction
- venous occlusion plethysmography – the measurement of changes in brachial artery volume and blood flow induced either by a mercury strain gauge during forearm hyperaemia stimulated by vasoactive agents, or reactive hyperaemia after forearm occlusion (figure 3).
Potential circulating biomarkers of endothelial dysfunction
These are:
- cellular adhesion molecules, e.g. vascular cell adhesion molecule-1 (VCAM-1), endothelial leukocyte adhesion molecule-1, E-selectin, intercellular adhesion molecule-1 (ICAM-1)
- von Willebrand factor
- endothelin -1
- bone marrow-derived endothelial progenitor cells.
Fatty streaks

A fatty streak marks the earliest macroscopic atherosclerotic lesion. Beginning in early life, these fatty streaks comprise a focal build-up of various substrates e.g. lipid-rich macrophages (foam cells), extracellular matrix, smooth muscle cells, intracellular and extracellular lipid deposits, and T lymphocytes, to form a minor thickening of the intimal surface.
The earliest detectable physiological manifestation of atherosclerosis is reduced production of the endothelium-dependent vasodilator NO2 (figure 1). Consequently, a vicious cycle develops whereby endothelial dysfunction and atherosclerosis enhance each other’s progression.

Fatty streaks (figure 4) may progress to become mature plaques.16,17 These can occur early in life and later develop to become advanced atherosclerotic lesions (figure 5).18 A low-saturated fat diet introduced in infancy and maintained during the first decade of life is associated with enhanced endothelial function in boys. This effect is explained partly by the diet-induced reduction in serum cholesterol concentration.18 Numerous studies have demonstrated the presence of fatty streaks and more advanced lesions with the progression of age. In a post-mortem study in the United States of 760 patients aged 15–34 years, who were victims of accidents, homicides or suicides, 2% of men aged 15–19 years and 20% of men aged 30–34 years had advanced lesions. Early atherosclerosis was associated with cardiovascular risk factors for coronary artery disease,19 with a preference for men compared to women. In vivo assessment of coronary arteries in 262 asymptomatic heart transplant patients one month after transplantation demonstrated a prevalence of atherosclerosis in 17% of patients under 20 years old, whilst those over 50 years had a prevalence of 85%. This study showed that atherosclerotic lesions are present in one in six teenagers.20
Plaque development

The progression of atherosclerotic plaques occurs through a number of processes (see reference 21 for an excellent review). These include:
- The accumulation of lipids within the lesion, in the form of acellular lipid-rich pools (extracellular cholesterol derived from LDL-cholesterol) or from the decay of lipid-containing macrophage foam cells.
- In early atherogenesis, atherogeneic lipoproteins are cleared by macrophages leaving intracellular lipid deposits. This results in foam cells and, as atherosclerosis progresses, this scavenging capacity is overwhelmed, which together with foam cell death, produces a lipid-rich core.
- The death of smooth muscle cells contributes to the accumulated lipid pools and furthers macrophage accumulation.
- A lipid-rich necrotic core develops within the lesion accompanied by inward remodelling (figure 6).
- Greater smooth muscle cell infiltration and the production of an extracellular matrix covered by a single layer of endothelial cells, produces a fibromuscular cap. This endothelium is defective, however, with increased permeability to particles e.g. lipoproteins and plasma molecules which extravasate into the subendothelial space.
- This overlies deeper extracellular lipid pools and is termed a fibrous plaque.
- Progressive macrophage infiltration, apoptosis (of endothelial, smooth muscle cells and macrophages) and hypoxia-induced necrosis promotes the development of advanced lesions. These contain necrotic lipid-rich cores and calcified regions and become the substrate for plaque complications and the development of clinical symptoms. The total amount of calcium (coronary artery calcium score) is a marker of coronary plaque burden.


Some plaques may cause severe stenoses (figure 7), while many remain angiographically invisible and begin to bulge into the adventitia rather than towards the lumen, a process referred to as Glagovian remodelling (figure 8). This is where the arterial media remodels to allow the vessel to increase its cross-sectional area and thereby accommodate the plaque without any reduction in luminal area (compensatory dilation).
The lipid content within plaques is dynamic with lipids being constantly transported in and out of atherosclerotic plaques. LDL and lipoprotein (a) particles contain cholesterol, triglycerides, phospholipids and apoliproteins, which can penetrate the endothelium. Taken up by arterial cells (endothelial cells, smooth muscle cells, macrophages or T lymphocytes), oxidation occurs to form oxidised LDL, which is particularly pro-atherogenic due to a variety of mechanisms:22
- as a chemoattractant for monocytes which develop into macrophages
- by promoting inflammation through macrophage cytokine release
- through intracellular uptake to form foam cells, which apoptose or rupture to release oxidised LDL, intracellular enzymes and oxygen free radicals
- by promoting endothelial dysfunction and reduced NO release
- acting as a pro-thrombotic to promote platelet aggregation and thromboxane release.
Plaque lipid content decreases after serum cholesterol levels reduce in animal models. Reverse cholesterol transport (also known as efflux) occurs predominantly through high-density lipoprotein (HDL) and macrophages – HDL is a heterogeneous population of particles in the blood, of 7-12nm in diameter, which can transport cholesterol from cholesterol-rich macrophages to the liver, where it is catabolised and eliminated. Consequently, it has anti-atherogenic properties. It is well-known from epidemiological studies that increased HDL levels are associated with a lower risk of cardiovascular disease.23,24 It has also been suggested that HDL can maintain endothelial function, protect against oxidation of LDL, and also has antithrombotic and anti-inflammatory effects.
Inflammation in atherosclerosis
Atherosclerosis has been described as a chronic inflammation of arteries, which develops over decades in response to the biological effects of risk factors.25
Inflammation plays a major role in all phases of atherosclerosis, a process recently reviewed by Bäck et al.26 Pro-inflammatory processes include intimal infiltration and oxidation of plasma-derived lipoproteins, their uptake by macrophages and subsequent development of foam cells. Within the plaque, hypoxia and inadequate removal of apoptotic and necrotic cell debris contribute to further inflammation and further atherogenesis. Conversely, an anti-inflammatory process that aims to resolve inflammation occurs simultaneously.
The balance between these two processes determines plaque stability and susceptibility to rupture. Inflammation-resolving lipid mediators are derived from omega-3 fatty acids or arachidonic acid and include other protein mediators such as annexin A1.26 Whereas pro-inflammatory mediators increase plaque size, core size and reduce cap thickness as well as plaque stability, pro-resolving mediators do the converse. Furthermore, pro-resolving mediators are important in the conversion of pro-inflammatory macrophages (M1 type) to anti-inflammatory macrophages (M2 type) that suppress inflammation and promote healing. Plaques with a net pro-inflammatory milieu are unstable and more prone to rupture; the opposite being true of a net pro-resolving process.
Studies to reduce inflammation, however, have been disappointing with no reduction in cardiovascular events seen. Elevated lipoprotein-associated phospholipase A2 (Lp-PLA2) activity promotes the development of atherosclerotic plaques, and elevated levels of this enzyme are associated with an increased risk of coronary events.27 The STABILITY study, however, found that the Lp-PLA2 inhibitor darapladib did not significantly reduce cardiovascular events in patients with stable coronary heart disease when added to usual standard of care.27 The SOLID-TIMI 52 study also found no reduction in coronary events in patients with ACS treated with darapladib.28
However, the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) demonstrated that a monoclonal antibody against interleukin-1β in patients with previous myocardial infarction and a high C-reactive protein (CRP) level (≥2mg/L) significantly reduced recurrent cardiovascular events independent of lipid lowering.29 Assessment of coronary inflammation is limited by the lack of a specific marker for the cardiovascular system (e.g. plasma biomarkers) or because tests are expensive and not readily available (e.g. hybrid positron emission tomography imaging). The use of perivascular fat attenuation index (FAI) on coronary computed tomography angiography (CCTA) may represent a novel assessment method.30 It captures changes in perivascular adipose tissue driven by inflammation – in a prospective cohort of patients with moderate to severe psoriasis, it was found that biologic therapy for psoriasis (e.g. anti-TNFα, anti-IL12/23/17) was associated with reduced coronary inflammation as assessed by perivascular FAI.31
Vulnerable plaque
The concept of rupture-prone (‘vulnerable’) plaques has arisen to encapsulate the biomechanical interplay between flow dynamics, plaque structure and biological processes that determine its mechanical strength.32 Plaque progression can occur in a number of ways through:
- plaque rupture in association with thin-cap atheroma
- plaque erosion in association with thicker fibrous caps, thrombus formation, and quiescent in terms of inflammation (which accounts for a quarter of ACS)
- calcified nodules disrupting plaque integrity and increasing the risk of plaque rupture.
Given this complex interplay, despite significant advancements in imaging allowing clinicians to assess rupture-prone plaques, risk prediction of acute coronary events has not improved compared with traditional approaches.33 It has been suggested that contemporary data do not support the vulnerable plaque concept and that thin-capped plaques do not inevitably rupture. In the Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT) study, only 5% of thin-capped plaques caused coronary events in 3.4 years of follow-up.34 Consequently, other mechanisms of ACS, e.g. plaque erosion, must be pursued and a greater focus placed on atherosclerotic disease burden rather than on features of individual plaques.35,36 The use of near-infrared spectroscopy (NIRS) intravascular ultrasound (IVUS) to quantify lipid core plaques and cholesterol content has recently been shown in the Lipid-Rich Plaque (LRP) study to identify vulnerable plaques and stratify patients at high risk of future cardiac events.33 It remains to be seen whether NIRS-guided therapy will mitigate future risk of adverse cardiac outcomes in these patients.
Major determinants of plaque vulnerability include both local and systemic factors.37 Local factors to the vulnerable plaque include:
- plaque burden
- size of lipid rich necrotic core
- thickness of the fibrous cap – a thin cap is more likely to break open (rupture or fissure) leading to thrombus formation during an ACS
- ongoing inflammation and repair within the cap
- eccentric (as distinct from concentric) plaque shape
- biomechanical forces including coronary tone and pressure, bending and twisting of the artery with cardiac contractility and coronary spasm.
Systemic factors promoting plaque rupture include:
- physical ‘triggers’ (e.g. exertion in normally sedentary individuals)
- high levels of psychological stress38 with attendant increased adrenergic tone, shear forces, and generalised inflammation
- diurnal variation in coagulation state
- autonomic vascular tone.
Patients are generally asymptomatic until plaque stenosis reaches 70–80% of lumen diameter when a reduction in flow is produced causing angina. Progression can either be chronic, with a slow narrowing and gradual accumulation of atheroma, or due to plaque haemorrhage, rupture and luminal thrombosis in the case of an ACS. Patients with thin-cap fibroatheroma are more likely to undergo plaque rupture, and repeated episodes of silent rupture; thrombosis and wound healing can increase plaque burden and stenosis.
The milieu of the plaque is also fundamental to its vulnerability. Evidence has implicated matrix metalloproteinases (MMPs) as major players in plaque rupture. MMPs are a complex group of proteinases with important roles in cardiovascular physiology and pathology.39 Their ability to degrade fibrous caps makes them an important target for future drug development for the prevention of ACS.
There continues to be much interest in the use of drugs (e.g. proprotein convertase subtilisin/kexin type 9 [PCSK9] inhibitors, canakinumab [monoclonal antibody targeting interleukin-1β]) that may modify the composition of these lesions to cause stabilisation and bring about a “quiescent coronary plaque”.16 In particular, statins have been shown to reduce the rate of progression as well as stabilise plaques that are prone to rupture.40 Evidence also suggests that statins have other non-lipid-lowering effects and can inhibit macrophage proliferation, reduce MMP and tissue factor (pro-thrombotic) expression by macrophages and increase inhibitors of metalloproteinase-1. Statins may also have anti-inflammatory effects, as they are associated with a reduction in serum CRP (elevated inflammatory markers, particularly CRP, are associated with atherosclerosis progression). Although not fully understood, other lipid-independent effects of statins include reversal of endothelial dysfunction, reduced thrombogenicity, decreased monocyte adhesion to the endothelium and reduced oxidation of LDL.
Biomarkers
Several studies have correlated different serologic biomarkers with cardiovascular disease. These biomarkers are useful since they can identify a population at risk of an acute ischaemic event and detect the presence of vulnerable plaques or vulnerable patients.
Ideally, a biomarker must have certain characteristics41 to predict the incidence of vascular disease. Measurements have to be reproducible in multiple independent samples, the method for determination should be standardised, variability should be controlled, and sensitivity and specificity should be high. In addition, the biomarker should add information to that provided by other, established risk markers and should reflect the underlying biologic process associated with plaque burden and progression.
Traditional biomarkers for cardiovascular risk include cigarette smoking, dyslipidaemia, hypertension and diabetes. While cardiovascular events can occur in patients who do not have these risk factors, in the majority of patients with coronary heart disease, these are likely to be present.42
C-reactive protein (CRP), although a non-specific but highly sensitive marker, may contribute to the pro-inflammatory state of the plaque by mediating monocyte recruitment and stimulating monocytes to release interleukin 1 (IL-1), IL-6, TNF-α.
Proposed biomarkers are listed below (although it should be noted that most are not in clinical use):
- inflammatory markers e.g. CRP, serum amyloid protein A, TNFα, myeloperoxidase (MPO), IL-6, Lp-PLA2
- imaging biomarker: e.g. FAI on CCTA
- markers of plaque erosion e.g. mast cells
- markers of thrombosis e.g. fibrinogen, thromboxane B2
- lipid-associated markers, e.g. high LDL-C (one of the strongest predictors of risk), oxLDL, or hypertriglyceridaemia
- markers of endothelial dysfunction – see above
- plaque stability e.g. MMPs. GDF-15
- metabolic markers e.g. HbA1c
- markers of neovascularisation, e.g. stroma-derived factor 1
- genetic markers e.g. gene polymorphisms of platelet glycoproteins and genes involved in cholesterol and lipoprotein metabolism, single nucleotide polymorphism (SNP) scores, such as LDL-C or polygenic, can identify patients with higher burden of atherosclerosis and greater response to therapy.43
Conclusion
A current challenge is to identify morphologic and molecular markers able to discriminate stable plaques from vulnerable ones, allowing the stratification of patients at high risk for acute cardiovascular events, including stroke, before clinical syndromes develop.41
close window and return to take test
References
- Fox KM. Exercise heart rate/ST segment relation. Perfect predictor of coronary disease. Br Heart J. 1982;48(4):309–10. http://dx.doi.org/10.1136/hrt.48.4.309
- Davies MJ, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–40. https://dx.doi.org/10.1056/NEJM198405033101801
- van der Wal AC, Becker AE. Atherosclerotic plaque rupture – pathologic basis of plaque stability and instability. Cardiovasc Res. 1999;41:334–44. https://dx.doi.org/10.1016/S0008-6363(98)00276-4
- Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction. A marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–75. http://dx.doi.org/10.1161/01.ATV.0000051384.43104.FC
- Ambrose JA, Barua RS, The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–7. https://dx.doi.org/10.1016/j.jacc.2003.12.047
- Messner B, Bernhard D. Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol 2014;34:509–15. https://dx.doi.org/10.1161/ATVBAHA.113.300156
- Ota Y, Kugiyama K, Sugiyama S et al. Impairment of endothelium-dependent relaxation of rabbit aortas by cigarette smoke extract – role of free radicals and attenuation by captopril. Atherosclerosis. 1997;131:195–202. https://dx.doi.org/10.1016/s0021-9150(97)06106-6
- Dikalov S, Itani H, Richmond B et al. Tobaccos smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension. Am J Physiol Heart Circ Physiol. 2019;316:H639–H646. https://dx.doi.org/10.1152/ajpheart.00595.2018
- Barua RS, Ambrose JA, Srivastava S, DeVoe MC, Eales-Reynolds RJ. Reactive oxygen species are involved in smoking-induced dysfunction of nitric oxide biosynthesis and upregulation of endothelial nitric oxide synthase: an in vitro demonstration in human coronary artery endothelial cells. Circulation. 2003;107:2342–7. http://circ.ahajournals.org/content/107/18/2342.full.pdf+html
- Lee WH, Ong SG, Zhou Y et al. Modeling cardiovascular risks of e-cigarettes with human-induced pluripotent stem cell-derived endothelial cells. J Am Coll Cardiol 2019;73:2722–37. https://dx.doi.org/10.1016/j.jacc.2019.03.476
- Rezk-Hanna M, Mosenifar Z, Benowitz NL et al. High carbon monoxide levels from charcoal combustion maks acute endothelial dysfunction induced by hookah (waterpipe) smoking in young adults. Circulation 2019;139:2215–24. https://dx.doi.org/10.1161/CIRCULATIONAHA.118.037375
- Mendall MA, Patel P, Asante M, et al. Relation of serum cytokine concentrations to cardiovascular risk factors and coronary heart disease. Heart 1997;78:273–7. http://dx.doi.org/10.1136/hrt.78.3.273
- Kianoush S, Yakoob MY, Al-Rifal M et al. Associations of cigarette smoking with subclinical inflammation and atherosclerosis: ELSA-Brasil (The Brazilian Longitudinal Study of Adult Health). J Am Heart Assoc 2017;6:e005088. https://dx.doi.org/10.1161/JAHA.116.005088
- Yokode M, Kita T, Arai H et al. Cholesteryl ester accumulation in macrophages incubated with low density lipoprotein pretreated with cigarette smoke extract. Proc Natl Acad Sci USA 1988;85:2344–8.
- Jezovnik MK. How to assess endothelial function for detection of pre-clinical atherosclerosis. Ejournal of Cardiology Practice 2011;10(10).
https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-10/How-to-assess-endothelial-function-for-detection-of-pre-clinical-atherosclerosis (last accessed 14th October 2019). - MacIsaac AI, Thomas JD, Topol EJ. Toward the quiescent coronary plaque. JACC 1993;22:1228–41. http://dx.doi.org/10.1016/0735-1097(93)90442-4
- Stary HC, Chandler AB, Glagov S et al. A definition initial, fatty streak, and intermediate lesions of atherosclerosis.Circulation 1994;89:2462–78. https://dx.doi.org/10.1161/01.CIR.89.5.2462
- Raitakari OT, Rönnemaa T, Järvisalo MJ et al. Endothelial function in healthy 11-year-old children after dietary intervention with onset in infancy; the Special Turku coronary Risk factor Intervention Project for Children (STRIP). Circulation 2005;112:3786–94. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.583195
- McGill HC Jr, McMahan CA, Zieske AW et al. Association of coronary heart disease risk factors with microscopic qualities of coronary atherosclerosis in youth. Circulation 2000;102:374–9. https://dx.doi.org/10.1161/01.cir.102.4.374
- Tuzcu EM, Kapadia SR, Tutar E et al. High prevalence of coronary atherosclerosis in asymptomatic teenagers and young adults: evidence from intravascular ultrasound. Circulation 2001;103:2705–10. https://dx.doi.org/10.1161/01.cir.103.22.2705
- Hansson G. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. https://dx.doi.org/10.1056/NEJMra043430
- Rosenson RS. Lipoprotein classification, metabolism, and role in atherosclerosis. https://www.uptodate.com/contents/lipoprotein-classification-metabolism-and-role-in-atherosclerosis (last accessed 14th October 2019)
- Miller NE. Associations of high-density lipoprotein subclasses and apolipoproteins with ischemic heart disease and coronary atherosclerosis. Am Heart J 1987;113:589–97. https://dx.doi.org/10.1016/0002-8703(87)90638-7
- Nicholls SJ, Nelson AJ. HDL and cardiovascular disease. Pathology 2019;51:142–7. https://dx.doi.org/10.1016/j.pathol.2018.10.017
- Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med 2012;366:54–63. http://dx.doi.org/10.1056/NEJMra1112570
- Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 2019;16:389–406. https://dx.doi.org/10.1038/s41569-019-0169-2
- The STABILITY investigators. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med 2014;370:1702−11. http://dx.doi.org/10.1056/NEJMoa1315878
- O’Donoghue ML, Braunwald E, White HD et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomised clinical trial. JAMA 2014;312:1006−15. http://dx.doi.org/10.1001/jama.2014.11061
- Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–31. http://dx.doi.org/10.1056/NEJMoa1707914
- Antoniades C, Antonopoulos AS, Deanfield J. Imaging residual inflammatory cardiovascular risk. Eur Heart J 2019 (published online 16th July 2019 ahead of print). http://dx.doi.org/10.1093/eurheartj/ehz474
- Elnabawi YA, Oikonomou EK, Dey AK et al. Association of biologic therapy with coronary inflammation with patients with psoriasis as assessed by perivascular fat attenuation index. JAMA Cardiol 2019 (published online 31st July 2019 ahead of print). http://dx.doi.org/10.1001/jamacardio.2019.2589
- Stefanadis C, Antoniou C-K, Tsiachris D, Pietri P. Coronary atherosclerotic vulnerable plaque: current perspectives. J Am Heart Assoc 2017;6:e005543 (published online 17th March 2017). http://dx.doi.org/10.1161/JAHA.117.005543
- Waksman R, Di Mario C, Torguson R et al. Identification of patients and plaques vulnerable to future coronary events with near-infrared spectroscopy intravascular ultrasound imaging: a prospective, cohort study. Lancet 2019; (published online 27th September 2019 ahead of print). http://doi.org/10.1016/S0140-6736(19)31794-5
- Stone GW, Maehara A, Lansky AJ et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med 2011;364:226–35. http://dx.doi.org/10.1056/NEJMoa1002358
- Arbab-Zadeh A, Fuster V. The myth of the “vulnerable plaque”: transitioning from a focus on individual lesions to atherosclertofic disease burden for coronary artery disease assessment. J Am Coll Cardiol 2015;65:846-55. http://dx.doi.org/10.1016/j.jacc.2014.11.041
- Libby P, Pasterkamp G. Requiem for the ‘vulnerable plaque’. Eur Heart J 2015;36:2984–7. http://doi.org/10.1093/eurheartj/ehv349
- Libby P. Molecular basis of the acute coronary syndromes. Circulation 1995;91:2844–50.
- Pignalberi C, Patti G, Chimenti C, et al. Role of different determinants of psychological distress in acute coronary syndromes. J Am Coll Cardiol 1998;32:613–19. http://dx.doi.org/10.1016/S0735-1097(98)00282-4
- Jones CB, Sane DC, Herrington DM. Matrix metalloproteinases: a review of their structure and role in acute coronary syndrome. Cardiovasc Res 2003;59:812–23. http://dx.doi.org/10.1016/S0008-6363(03)00516-9
- Nilsson J. Atherosclerotic plaque vulnerability in the statin era. Eur Heart J 2017;38:1638–44. https://dx.doi.org/10.1093/eurheartj/ehx143
- Spagnoli LG, Bonanno E, Sangiorgi G, Mauriello A. Role of inflammation in atherosclerosis. J Nucl Med 2007;48:1800–15. http://dx.doi.org/10.2967/jnumed.107.038661
- Tsimikas S, Willerson JT, Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol 2006;47(suppl 8):C19–C31. http://dx.doi.org/10.1016/j.jacc.2005.10.066
- Natarajan P, Young R, Stitziel NO et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 2017;135:2091-101. https://dx.doi.org/10.1161/CIRCULATIONAHA.116.024436

All rights reserved. No part of this programme may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without the prior permission of the publishers, Medinews (Cardiology) Limited.
It shall not, by way of trade or otherwise, be lent, re-sold, hired or otherwise circulated without the publisher’s prior consent.
Medical knowledge is constantly changing. As new information becomes available, changes in treatment, procedures, equipment and the use of drugs becomes necessary. The editors/authors/contributors and the publishers have taken care to ensure that the information given in this text is accurate and up to date. Readers are strongly advised to confirm that the information, especially with regard to drug usage, complies with the latest legislation and standards of practice.
Healthcare professionals should consult up-to-date Prescribing Information and the full Summary of Product Characteristics available from the manufacturers before prescribing any product. Medinews (Cardiology) Limited cannot accept responsibility for any errors in prescribing which may occur.